Fraunhofer Institute for Toxicology and Experimental Medicine, Hannover, Germany.
Department of Respiratory Medicine, University Medical Center, Freiburg, Germany.
Front Immunol. 2021 Apr 23;12:642855. doi: 10.3389/fimmu.2021.642855. eCollection 2021.
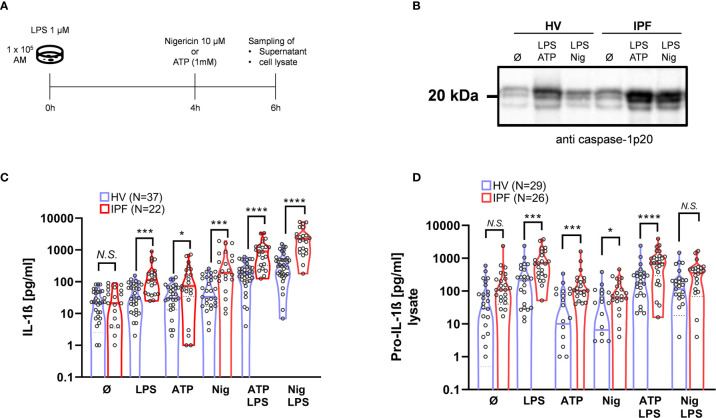
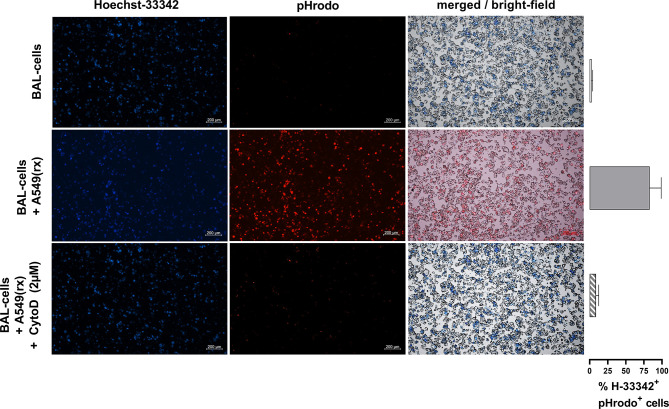
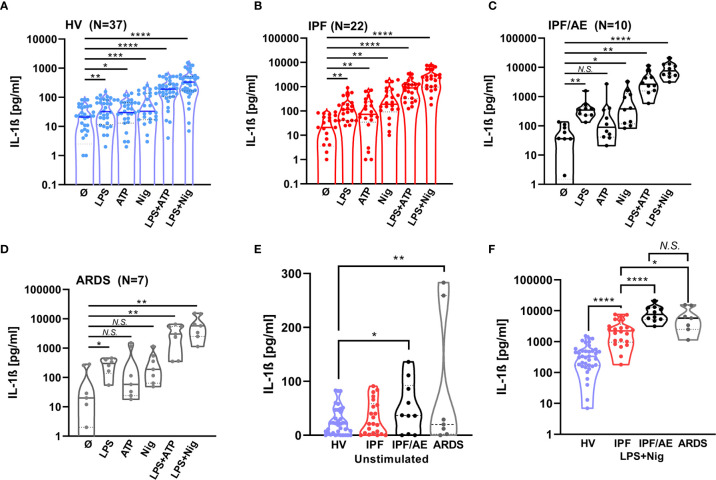
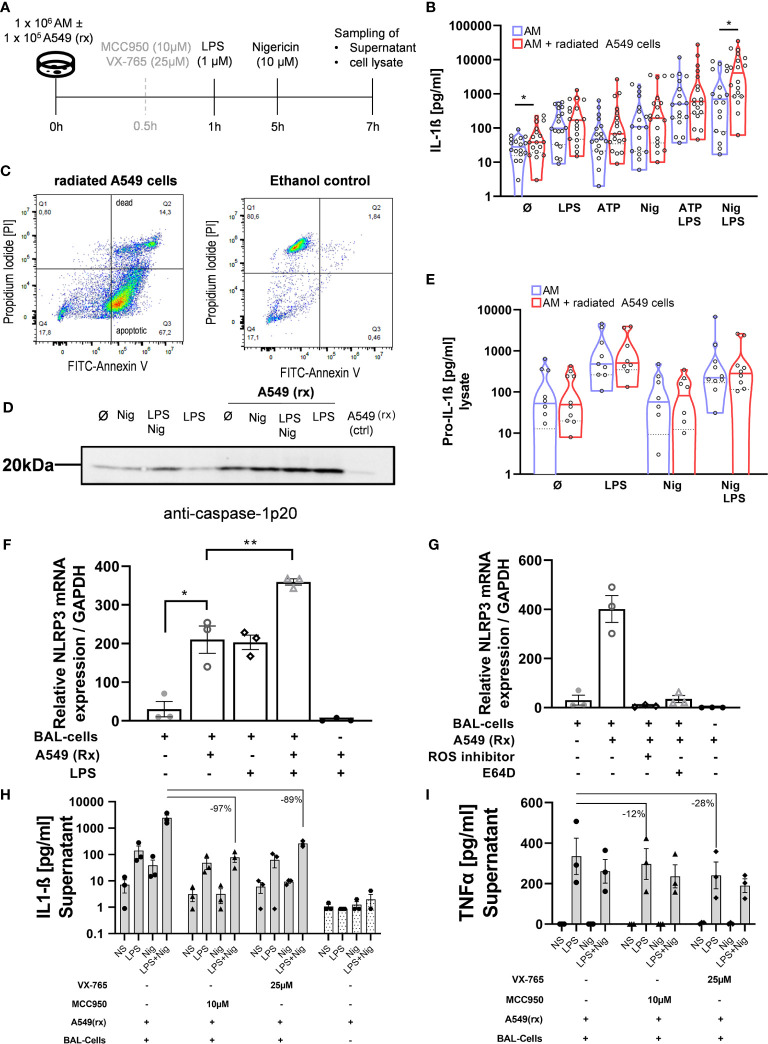
Idiopathic pulmonary fibrosis (IPF) is a relentlessly progressive disease harboring significant morbidity and mortality despite recent advances in therapy. Regardless of disease severity acute exacerbations (IPF-AEs) may occur leading to considerable loss of function and are the leading cause of death in IPF. Histologic features of IPF-AE are very similar to acute respiratory distress syndrome (ARDS), but the underlying mechanisms are incompletely understood. We investigated the role of the NLRP3 inflammasome in IPF and IPF-AE. Bronchoalveolar lavage (BAL) cells were sampled from patients with IPF (n = 32), IPF-AE (n = 10), ARDS (n = 7) and healthy volunteers (HV, n = 37) and the NLRP3-inflammasome was stimulated . We found the NLRP3 inflammasome to be hyper-inducible in IPF compared to HV with increased IL-1ß and pro-IL-1ß levels on ELISA upon stimulation as well as increased caspase-1 activity measured by caspase-1p20 immunoblotting. In IPF-AE, IL-1ß was massively elevated to an extent similar to ARDS. To evaluate potential mechanisms, we co-cultured BAL cells with radiated A549 cells (a model to simulate apoptotic alveolar epithelial cells), which led to increased NLRP3 mRNA expression and increased caspase-1 dependent IL-1ß production. In the presence of a reactive oxygen species (ROS) inhibitor (diphenyleneiodonium) and a cathepsin B inhibitor (E64D), NLRP3 expression was suppressed indicating that induction of NLRP3 activation following efferocytosis of apoptotic A549 cells is mediated ROS and cathepsin-B. In summary, we present evidence of involvement of the NLRP3 inflammasome-caspase pathway in the pathogenesis of IPF-AE, similarly to ARDS, which may be mediated by efferocytosis of apoptotic alveolar epithelial cells in IPF.
特发性肺纤维化(IPF)是一种进行性疾病,尽管治疗取得了最近的进展,但仍存在显着的发病率和死亡率。无论疾病严重程度如何,急性加重(IPF-AE)都可能发生,导致功能大量丧失,是 IPF 的主要死亡原因。IPF-AE 的组织学特征与急性呼吸窘迫综合征(ARDS)非常相似,但潜在机制尚不完全清楚。我们研究了 NLRP3 炎性体在 IPF 和 IPF-AE 中的作用。从特发性肺纤维化患者(n=32)、特发性肺纤维化急性加重患者(n=10)、ARDS 患者(n=7)和健康志愿者(HV,n=37)中采集支气管肺泡灌洗液(BAL)细胞,并刺激 NLRP3 炎性体。我们发现,与 HV 相比,IPF 中的 NLRP3 炎性体具有更高的诱导性,刺激后 ELISA 检测到 IL-1β 和 pro-IL-1β 水平升高,以及 caspase-1p20 免疫印迹检测到 caspase-1 活性升高。在 IPF-AE 中,IL-1β 大量升高,程度与 ARDS 相似。为了评估潜在的机制,我们将 BAL 细胞与辐射 A549 细胞共培养(模拟凋亡肺泡上皮细胞的模型),这导致 NLRP3 mRNA 表达增加和 caspase-1 依赖性 IL-1β 产生增加。在活性氧(ROS)抑制剂(二苯乙烯碘)和组织蛋白酶 B 抑制剂(E64D)存在的情况下,NLRP3 表达受到抑制,表明凋亡 A549 细胞吞噬作用后 NLRP3 激活的诱导是由 ROS 和组织蛋白酶 B 介导的。总之,我们提供了证据表明 NLRP3 炎性体-胱天蛋白酶途径参与了 IPF-AE 的发病机制,与 ARDS 相似,这可能是由 IPF 中凋亡肺泡上皮细胞的吞噬作用介导的。