Singh Nitya, Li Xiaolong, Beshearse Elizabeth, Blanton Jason L, DeMent Jamie, Havelaar Arie H
Animal Sciences Department, Emerging Pathogens Institute, Food Systems Institute, University of Florida, Gainesville, FL, United States.
Department of Environmental and Global Health, Emerging Pathogens Institute, University of Florida, Gainesville, FL, United States.
Front Med (Lausanne). 2021 Apr 22;8:656827. doi: 10.3389/fmed.2021.656827. eCollection 2021.
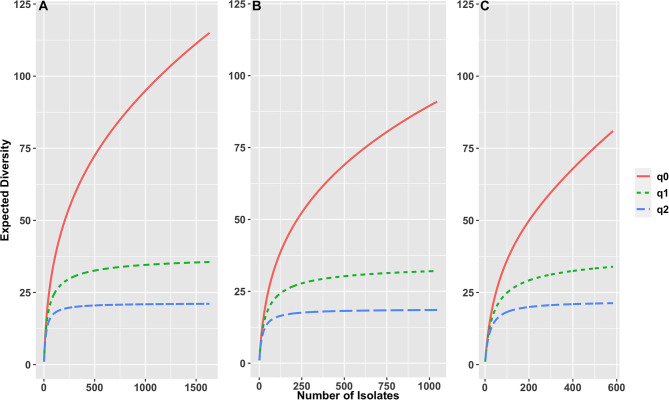
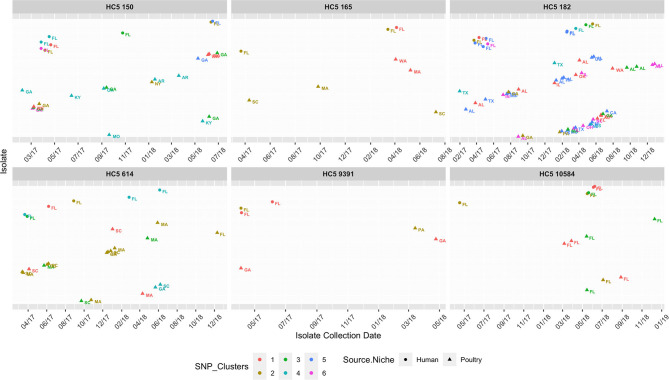
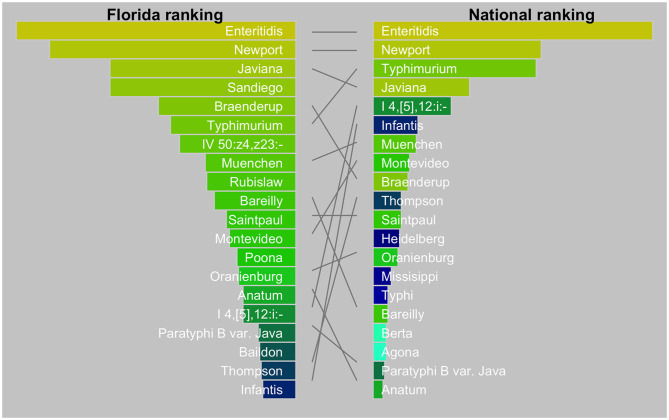
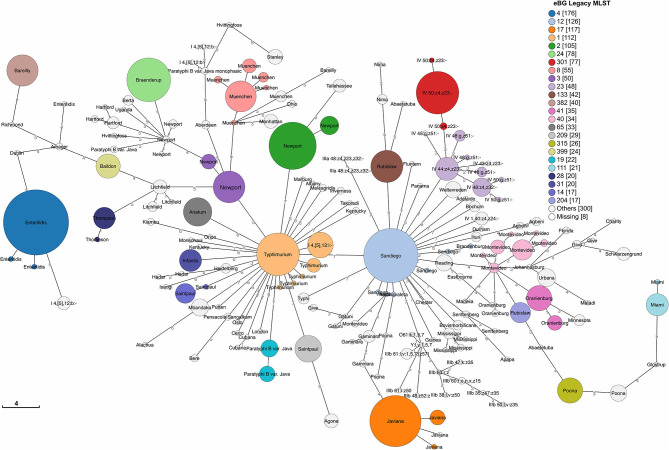
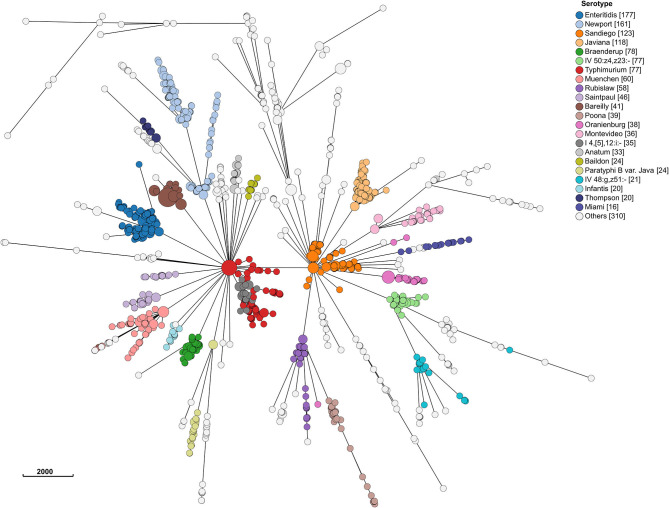
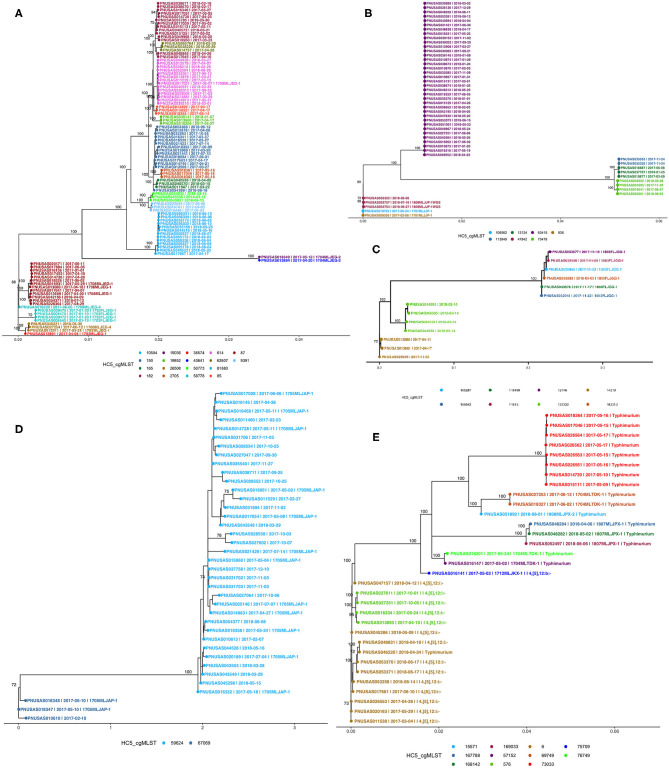
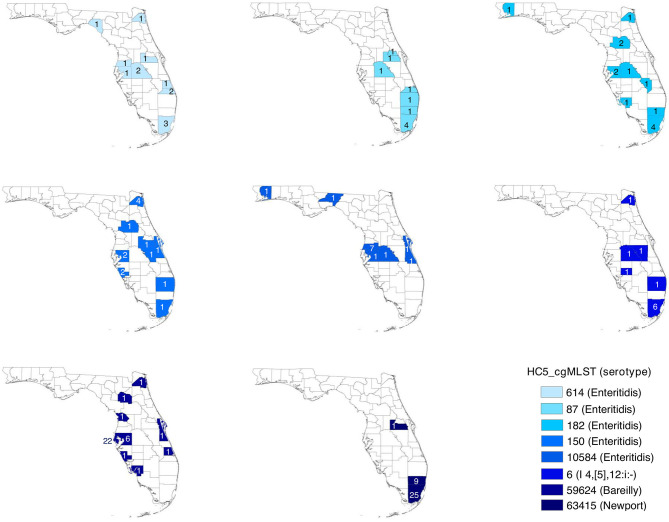
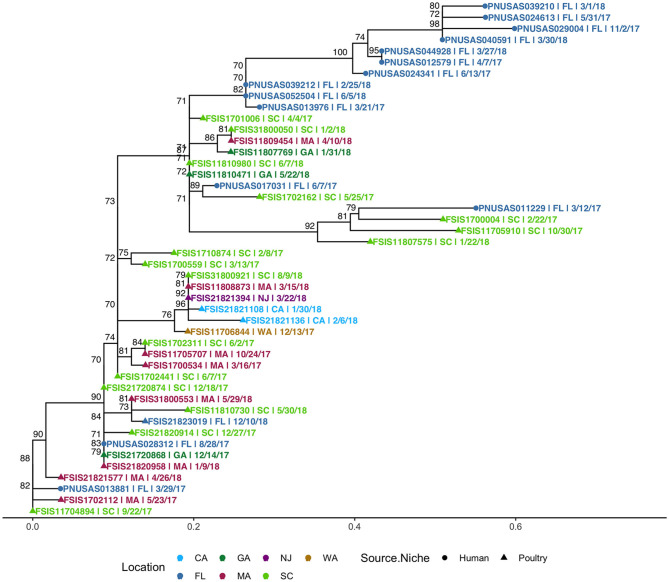
The state of Florida reports a high burden of non-typhoidal with approximately two times higher than the national incidence. We retrospectively analyzed the population structure and molecular epidemiology of 1,709 clinical isolates from 2017 and 2018. We found 115 different serotypes. Rarefaction suggested that the serotype richness did not differ between children under 2 years of age and older children and adults and, there are ~22 well-characterized dominant serotypes. There were distinct differences in dominant serotypes between Florida and the USA as a whole, even though . Enteritidis and . Newport were the dominant serotypes in Florida and nationally. . Javiana, . Sandiego, and . IV 50:z4, z23:- occurred more frequently in Florida than nationally. Legacy Multi Locus Sequence Typing (MLST) was of limited use for differentiating clinical isolates beyond the serotype level. We utilized core genome MLST (cgMLST) hierarchical clusters (HC) to identify potential outbreaks and compared them to outbreaks detected by Pulse Field Gel Electrophoresis (PFGE) surveillance for five dominant serotypes (Enteritidis, Newport, Javiana, Typhimurium, and Bareilly). Single nucleotide polymorphism (SNP) phylogenetic-analysis of cgMLST HC at allelic distance 5 or less (HC5) corroborated PFGE detected outbreaks and generated well-segregated SNP distance-based clades for all studied serotypes. We propose "combination approach" comprising " as efficient tool to trigger outbreak investigations, and " for higher resolution phylogeny to confirm an outbreak. We also applied this approach to identify case clusters, more distant in time and place than traditional outbreaks but may have been infected from a common source, comparing 176 Florida clinical isolates and 1,341 non-clinical isolates across USA, of most prevalent serotype Enteritidis collected during 2017-2018. Several clusters of closely related isolates (0-4 SNP apart) within HC5 clusters were detected and some included isolates from poultry from different states in the US, spanning time periods over 1 year. Two SNP-clusters within the same HC5 cluster included isolates with the same multidrug-resistant profile from both humans and poultry, supporting the epidemiological link. These clusters likely reflect the vertical transmission of clones from higher levels in the breeding pyramid to production flocks.
佛罗里达州报告非伤寒性负担较高,发病率约为全国发病率的两倍。我们回顾性分析了2017年和2018年1709株临床分离株的种群结构和分子流行病学。我们发现了115种不同的血清型。稀疏分析表明,2岁以下儿童与大龄儿童及成人之间的血清型丰富度没有差异,并且有大约22种特征明确的优势血清型。尽管肠炎沙门氏菌和纽波特沙门氏菌在佛罗里达州和全国都是优势血清型,但佛罗里达州与美国整体的优势血清型存在明显差异。哈维亚纳沙门氏菌、圣地亚哥沙门氏菌和IV 50:z4,z23:-在佛罗里达州比在全国更为常见。传统多位点序列分型(MLST)在血清型水平之外区分临床分离株的作用有限。我们利用核心基因组MLST(cgMLST)层次聚类(HC)来识别潜在的疫情爆发,并将其与通过脉冲场凝胶电泳(PFGE)监测检测到的五种优势血清型(肠炎沙门氏菌、纽波特沙门氏菌、哈维亚纳沙门氏菌、鼠伤寒沙门氏菌和巴雷利沙门氏菌)的疫情爆发进行比较。等位基因距离为5或更小的cgMLST HC的单核苷酸多态性(SNP)系统发育分析证实了PFGE检测到的疫情爆发,并为所有研究的血清型生成了基于SNP距离的良好分离的进化枝。我们提出“组合方法”,包括将“作为触发疫情调查的有效工具,以及将“用于更高分辨率的系统发育分析以确认疫情爆发。我们还应用这种方法来识别病例集群,这些病例集群在时间和地点上比传统疫情爆发更为遥远,但可能来自共同来源,比较了2017 - 2018年期间收集的佛罗里达州176株临床分离株和美国各地1341株非临床分离株中最常见的血清型肠炎沙门氏菌。在HC5集群中检测到几个密切相关的分离株集群(等位基因距离为0 - 4个SNP),其中一些包括来自美国不同州的家禽分离株,跨越时间超过1年。同一HC5集群内的两个SNP集群包括来自人类和家禽的具有相同多重耐药谱的分离株,支持了流行病学联系。这些集群可能反映了克隆从育种金字塔较高层次向生产鸡群的垂直传播。