Section of Pulmonary & Critical Care Medicine, VA Portland Health Care System, Portland, United States.
Department of Medicine, Division of Pulmonary & Critical Care Medicine, Oregon Health & Science University, Portland, United States.
Elife. 2021 May 11;10:e67914. doi: 10.7554/eLife.67914.
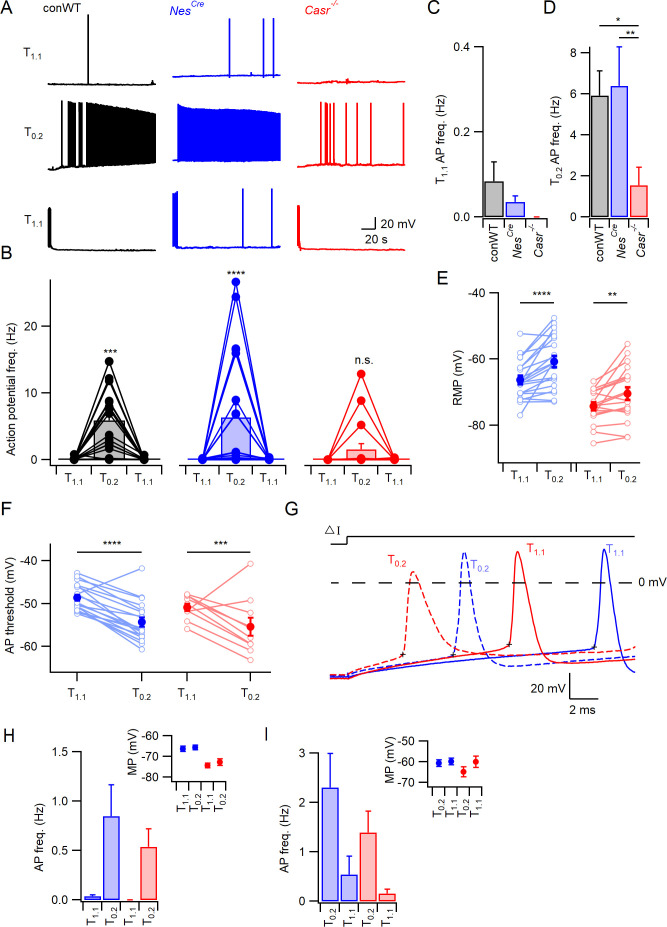
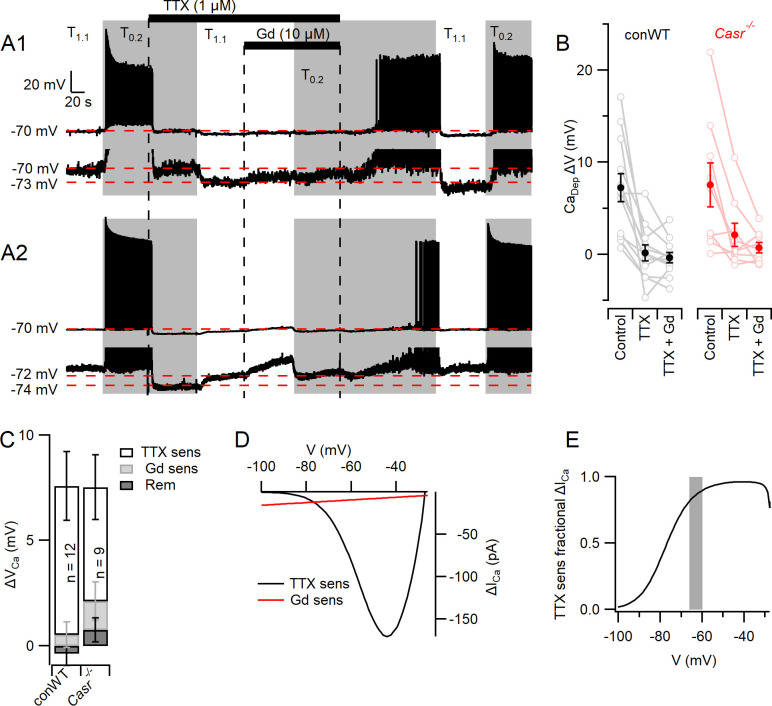
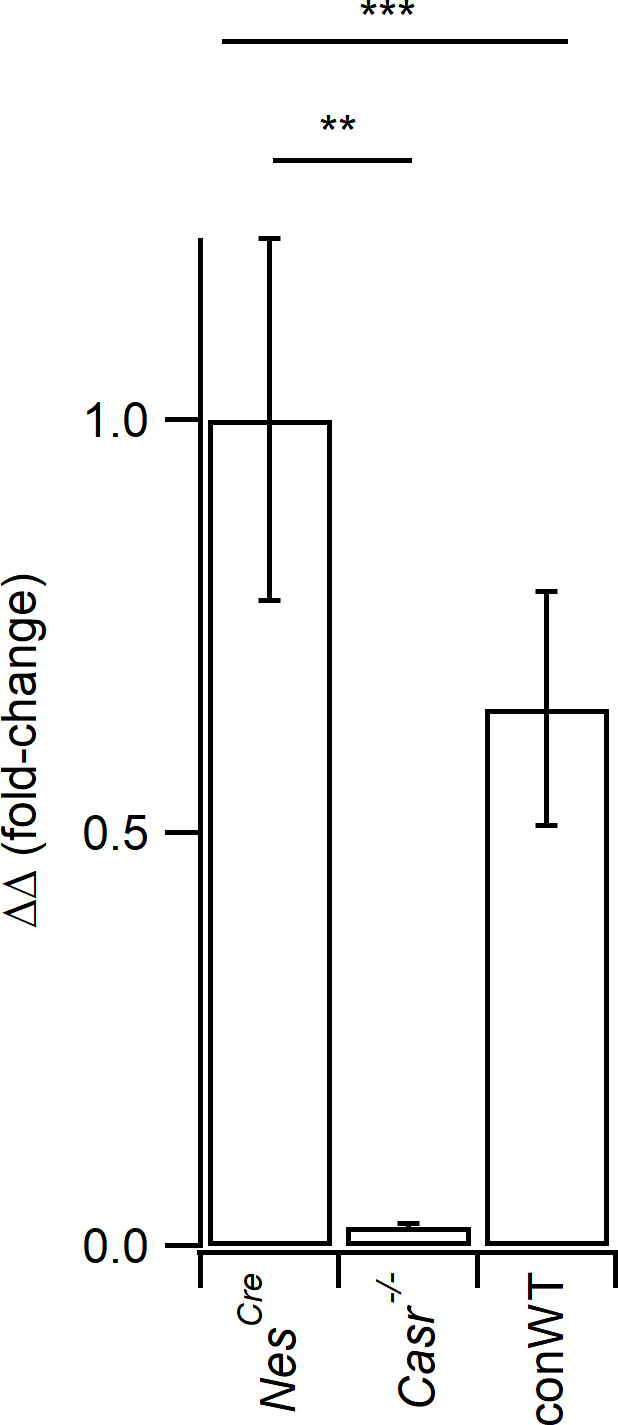
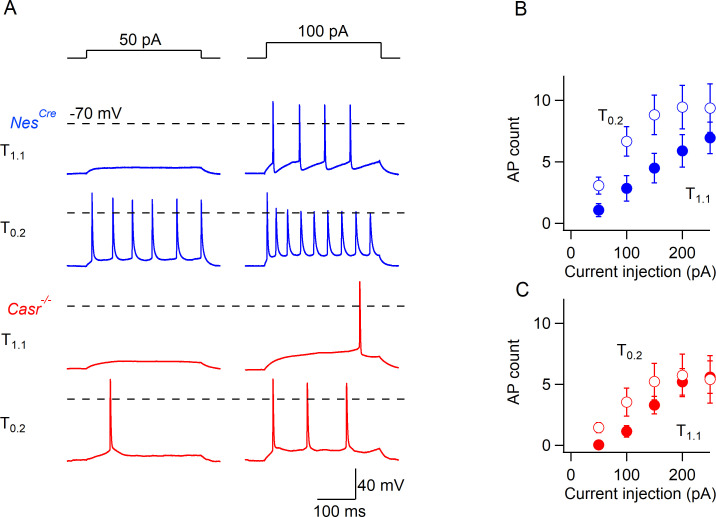
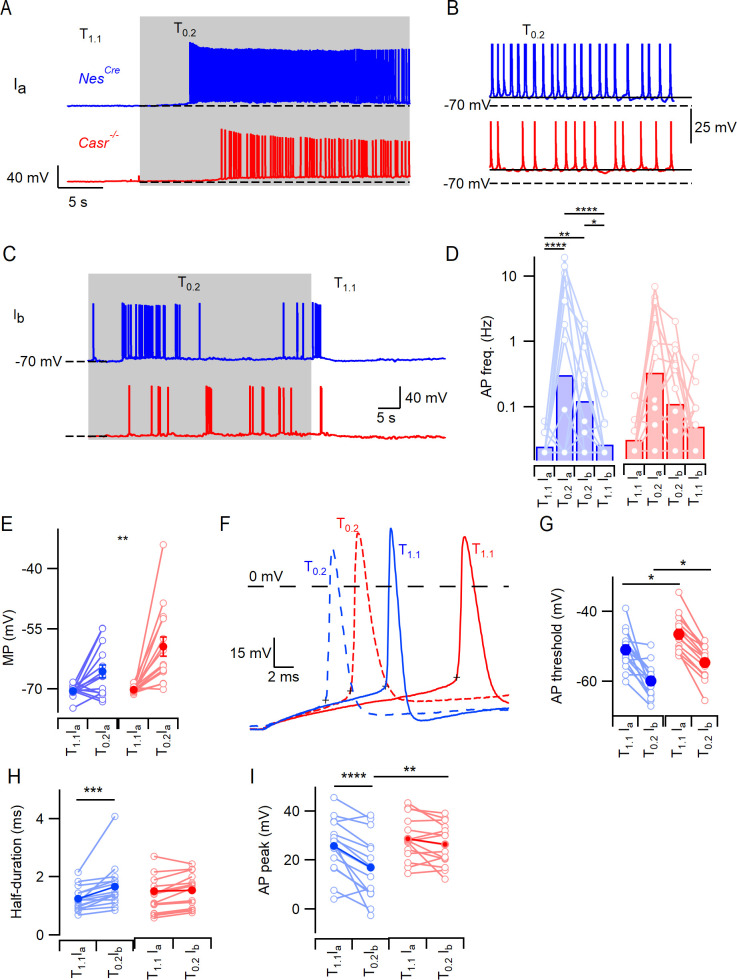
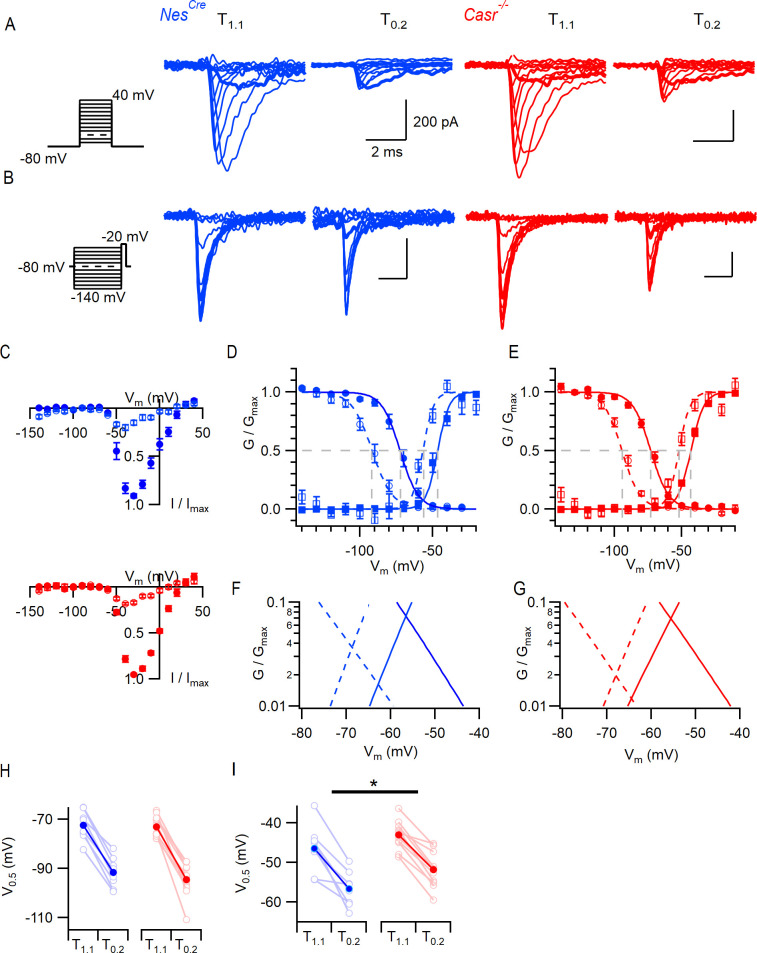
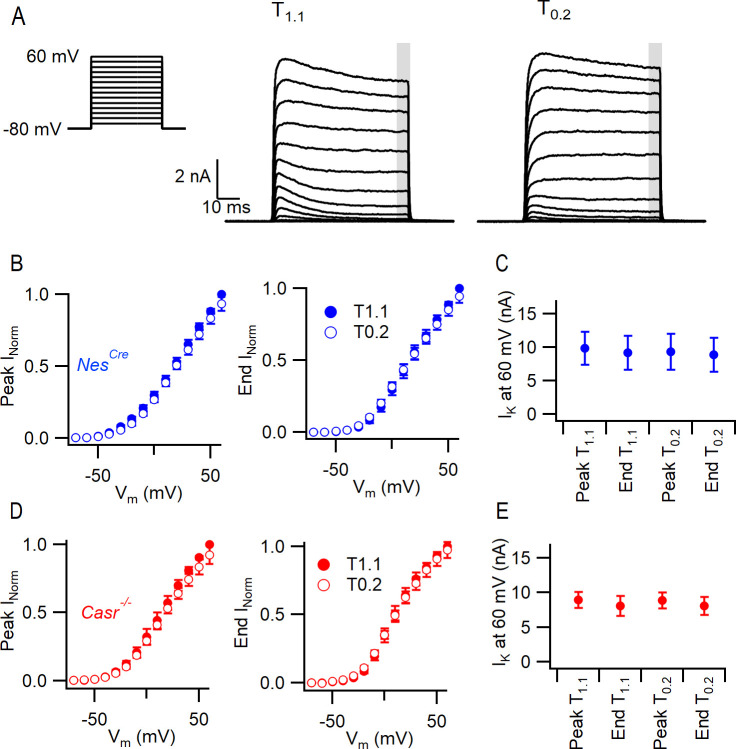
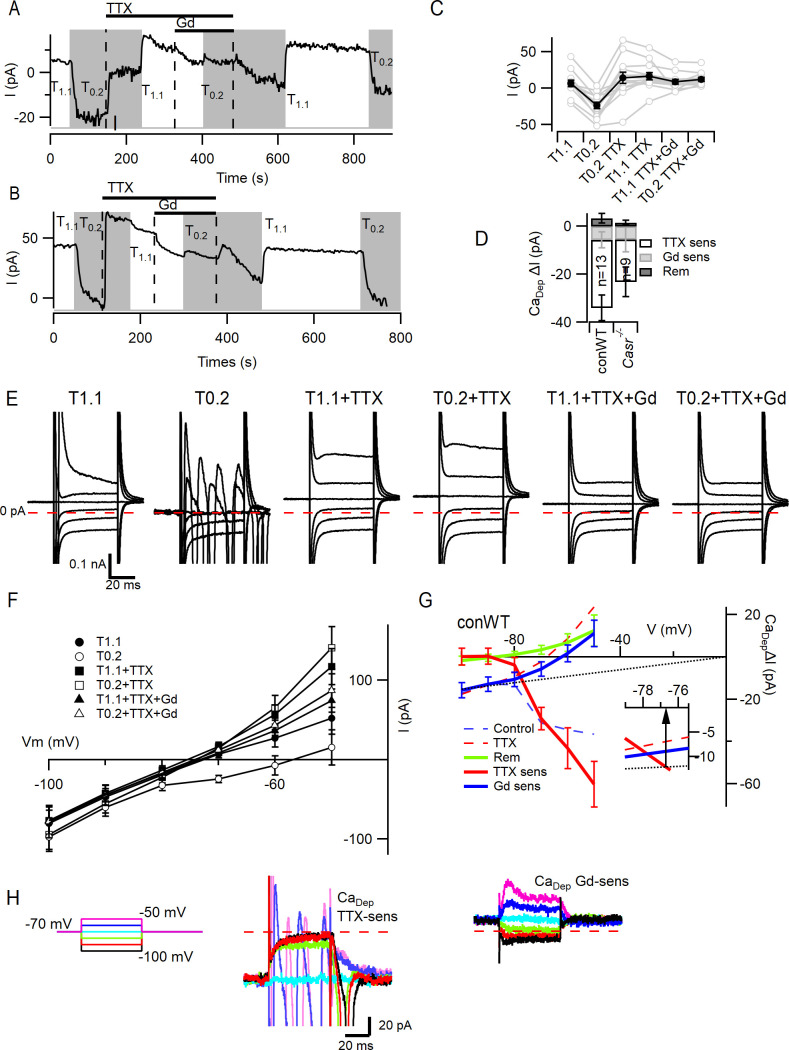
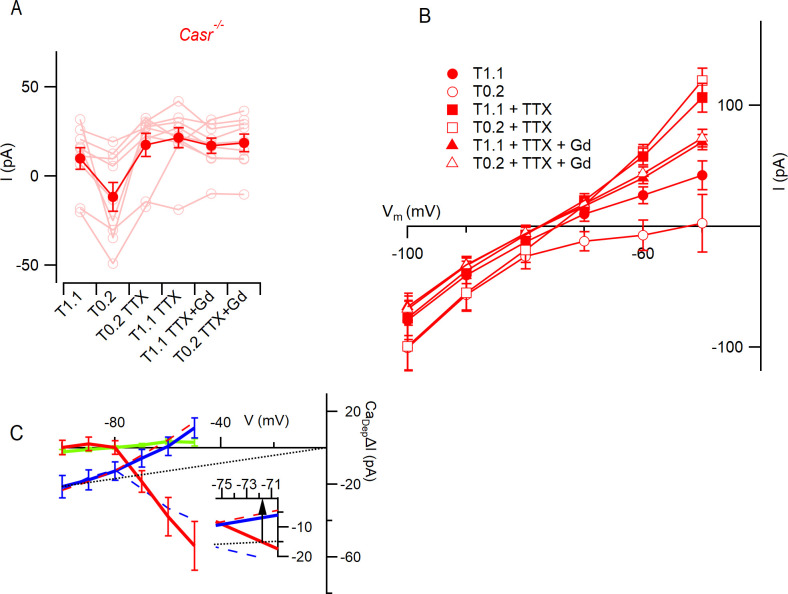
Increasing extracellular [Ca2+] ([Ca2+]o) strongly decreases intrinsic excitability in neurons but the mechanism is unclear. By one hypothesis, [Ca2+]o screens surface charge, reducing voltage-gated sodium channel (VGSC) activation and by another [Ca2+]o activates Calcium-sensing receptor (CaSR) closing the sodium-leak channel (NALCN). Here we report that neocortical neurons from CaSR-deficient (Casr-/-) mice had more negative resting potentials and did not fire spontaneously in reduced divalent-containing solution (T0.2) in contrast with wild-type (WT). However, after setting membrane potential to -70 mV, T0.2 application similarly depolarized and increased action potential firing in Casr-/- and WT neurons. Enhanced activation of VGSCs was the dominant contributor to the depolarization and increase in excitability by T0.2 and occurred due to hyperpolarizing shifts in VGSC window currents. CaSR deletion depolarized VGSC window currents but did not affect NALCN activation. Regulation of VGSC gating by external divalents is the key mechanism mediating divalent-dependent changes in neocortical neuron excitability.
细胞外钙离子浓度 ([Ca2+]o) 的增加强烈降低神经元的固有兴奋性,但机制尚不清楚。根据一种假说,[Ca2+]o 屏蔽表面电荷,从而降低电压门控钠离子通道 (VGSC) 的激活,而另一种假说则认为 [Ca2+]o 激活钙敏感受体 (CaSR) 关闭钠漏通道 (NALCN)。在这里,我们报告说,来自钙敏感受体缺失 (Casr-/-) 小鼠的新皮层神经元在低二价阳离子含量溶液 (T0.2) 中具有更负的静息电位,并且不会自发放电,而野生型 (WT) 则相反。然而,将膜电位设置为 -70 mV 后,T0.2 的应用同样使 Casr-/- 和 WT 神经元去极化并增加动作电位的发放。VGSC 的激活增强是 T0.2 去极化和兴奋性增加的主要贡献者,这是由于 VGSC 窗口电流的超极化偏移引起的。CaSR 的缺失使 VGSC 窗口电流去极化,但不影响 NALCN 的激活。外部二价离子对 VGSC 门控的调节是介导新皮层神经元兴奋性中二价依赖性变化的关键机制。