Department of Molecular Genetics, The Weizmann Institute of Science, Rehovot 76100, Israel.
Department of Immunology, The Weizmann Institute of Science, Rehovot 76100, Israel.
J Cell Sci. 2021 May 1;134(9). doi: 10.1242/jcs.254979. Epub 2021 May 11.
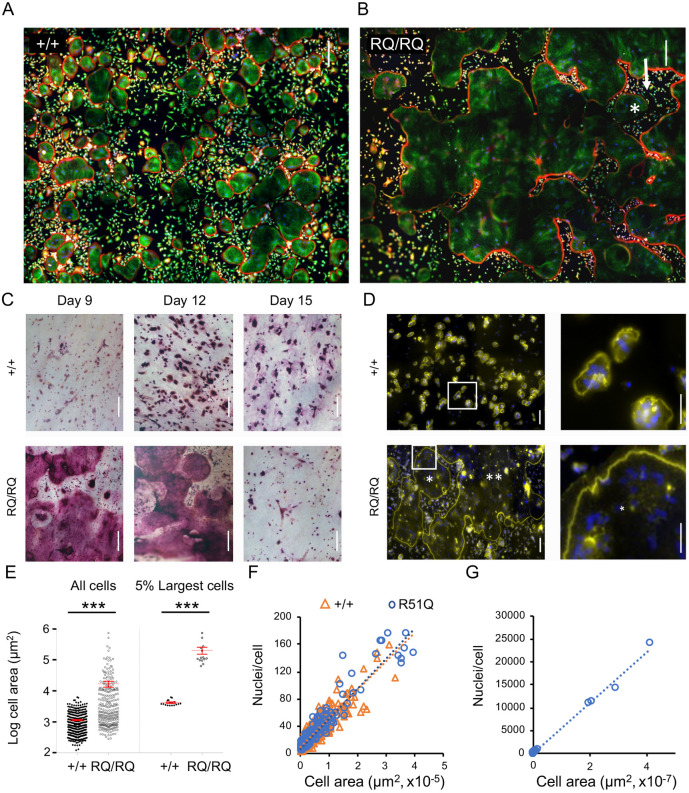
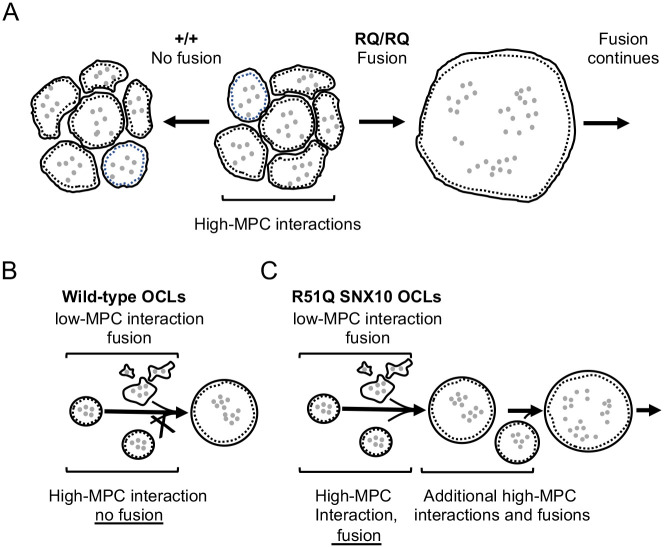
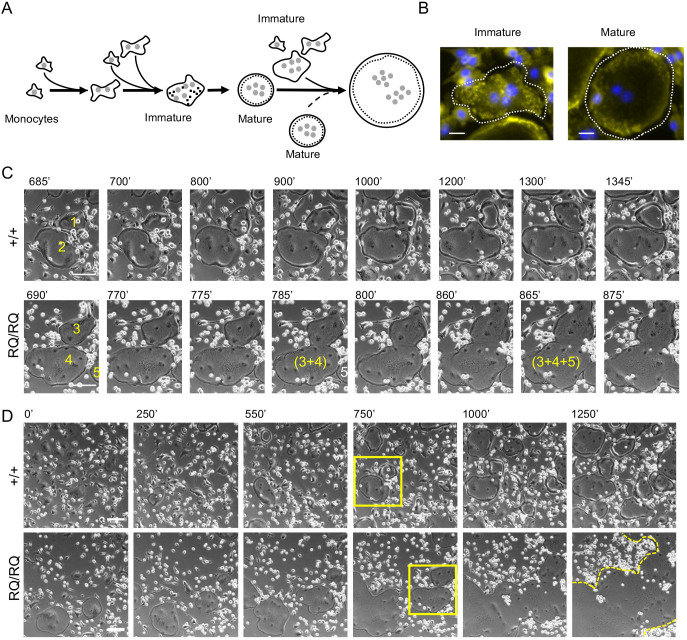
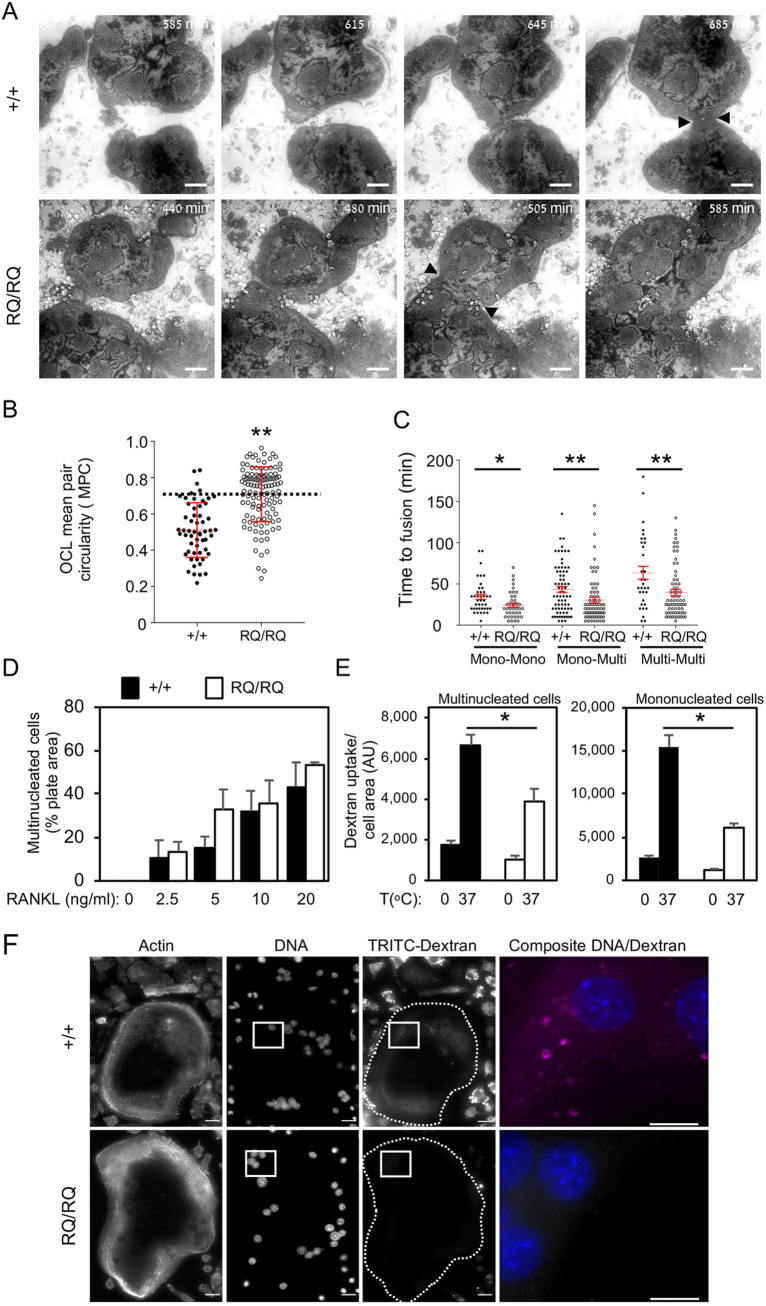
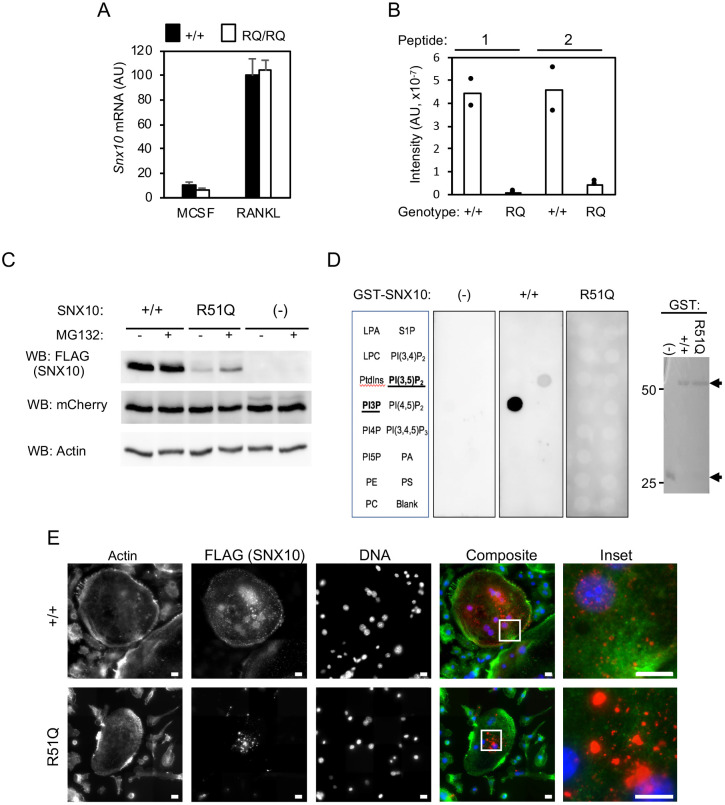
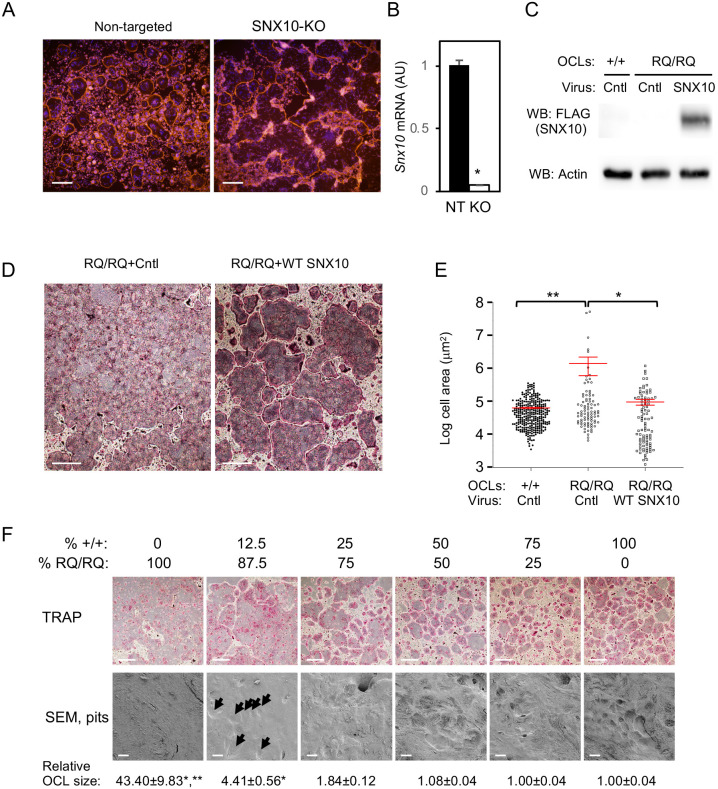
Homozygosity for the R51Q mutation in sorting nexin 10 (SNX10) inactivates osteoclasts (OCLs) and induces autosomal recessive osteopetrosis in humans and in mice. We show here that the fusion of wild-type murine monocytes to form OCLs is highly regulated, and that its extent is limited by blocking fusion between mature OCLs. In contrast, monocytes from homozygous R51Q SNX10 mice fuse uncontrollably, forming giant dysfunctional OCLs that can become 10- to 100-fold larger than their wild-type counterparts. Furthermore, mutant OCLs display reduced endocytotic activity, suggesting that their deregulated fusion is due to alterations in membrane homeostasis caused by loss of SNX10 function. This is supported by the finding that the R51Q SNX10 protein is unstable and exhibits altered lipid-binding properties, and is consistent with a key role for SNX10 in vesicular trafficking. We propose that OCL size and functionality are regulated by a cell-autonomous SNX10-dependent mechanism that downregulates fusion between mature OCLs. The R51Q mutation abolishes this regulatory activity, leading to excessive fusion, loss of bone resorption capacity and, consequently, to an osteopetrotic phenotype in vivo. This article has an associated First Person interview with the joint first authors of the paper.
SNX10 中的 R51Q 突变纯合导致破骨细胞(OCL)失活,并在人类和小鼠中诱导常染色体隐性骨硬化症。我们在这里表明,野生型小鼠单核细胞融合形成破骨细胞受到高度调控,其程度受到成熟破骨细胞之间融合的阻断限制。相比之下,来自 R51Q SNX10 纯合子小鼠的单核细胞不受控制地融合,形成巨大的功能失调的破骨细胞,其大小可以是野生型细胞的 10 到 100 倍。此外,突变型破骨细胞显示出降低的内吞活性,表明其不受控制的融合是由于 SNX10 功能丧失导致的膜稳态改变所致。这一发现得到了支持,即 R51Q SNX10 蛋白不稳定,并表现出改变的脂质结合特性,这与 SNX10 在囊泡运输中的关键作用一致。我们提出,破骨细胞的大小和功能受到一种细胞自主的、依赖 SNX10 的机制的调节,该机制下调成熟破骨细胞之间的融合。R51Q 突变消除了这种调节活性,导致过度融合、丧失骨吸收能力,进而导致体内出现骨硬化表型。本文附有该论文的共同第一作者的相关第一人称采访。