School of Physics, The University of Sydney, Camperdown, NSW, Australia.
The Turner Institute for Brain and Mental Health, School of Psychological Sciences and Monash Biomedical Imaging, Monash University, Clayton, VIC, Australia.
Nat Commun. 2021 May 11;12(1):2669. doi: 10.1038/s41467-021-22862-1.
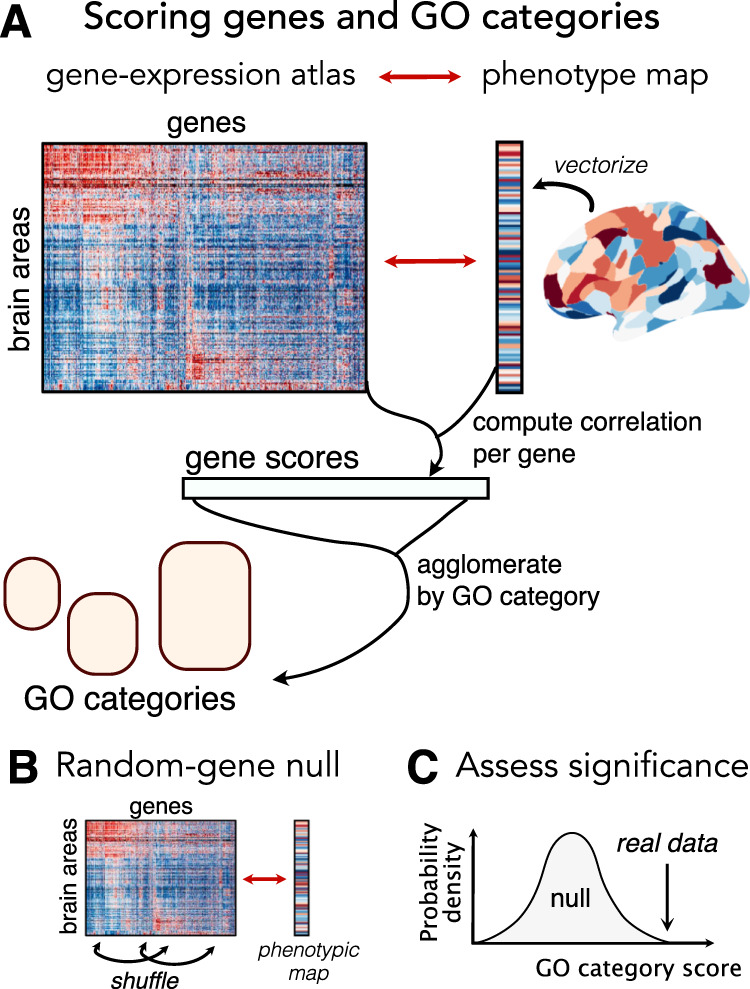
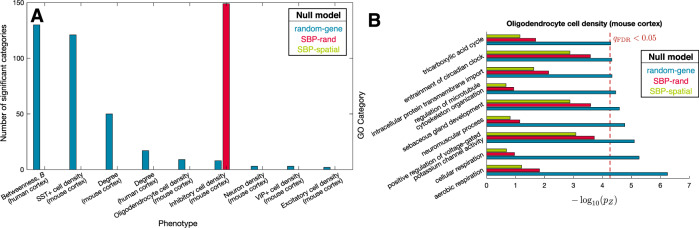
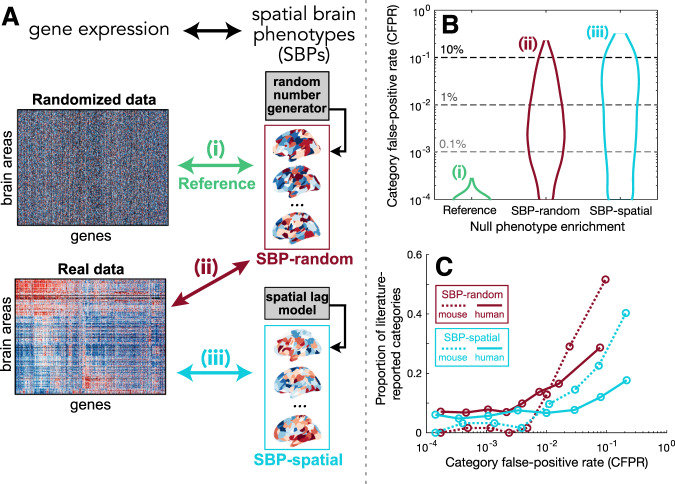
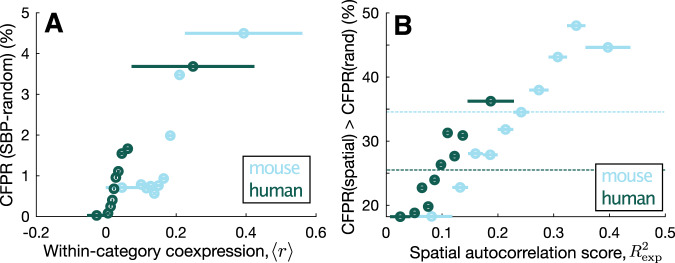
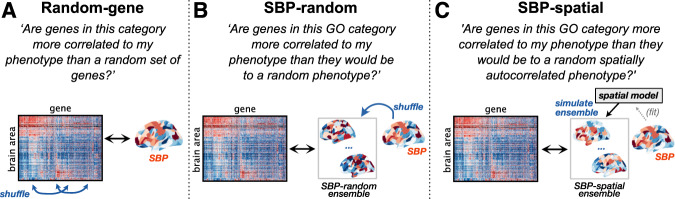
Transcriptomic atlases have improved our understanding of the correlations between gene-expression patterns and spatially varying properties of brain structure and function. Gene-category enrichment analysis (GCEA) is a common method to identify functional gene categories that drive these associations, using gene-to-category annotation systems like the Gene Ontology (GO). Here, we show that applying standard GCEA methodology to spatial transcriptomic data is affected by substantial false-positive bias, with GO categories displaying an over 500-fold average inflation of false-positive associations with random neural phenotypes in mouse and human. The estimated false-positive rate of a GO category is associated with its rate of being reported as significantly enriched in the literature, suggesting that published reports are affected by this false-positive bias. We show that within-category gene-gene coexpression and spatial autocorrelation are key drivers of the false-positive bias and introduce flexible ensemble-based null models that can account for these effects, made available as a software toolbox.
转录组图谱提高了我们对基因表达模式与大脑结构和功能的空间变化特性之间相关性的理解。基因类别富集分析(GCEA)是一种常用的方法,用于识别驱动这些关联的功能基因类别,使用基因到类别注释系统,如基因本体论(GO)。在这里,我们表明,将标准 GCEA 方法应用于空间转录组数据会受到大量假阳性偏差的影响,GO 类别与随机神经表型的假阳性关联的平均膨胀超过 500 倍,在小鼠和人类中。GO 类别的估计假阳性率与其在文献中被报道为显著富集的频率相关,这表明已发表的报告受到这种假阳性偏差的影响。我们表明,类别内基因-基因共表达和空间自相关是假阳性偏差的关键驱动因素,并引入了灵活的基于集成的零模型,可以解释这些影响,作为软件工具箱提供。