Department of Pediatrics, Division of Allergy Immunology, Rady Children's Hospital, University of California San Diego, San Diego, CA, USA.
Trinity Life Sciences, Waltham, MA, USA.
J Clin Immunol. 2021 Jul;41(5):881-895. doi: 10.1007/s10875-021-01059-7. Epub 2021 May 13.
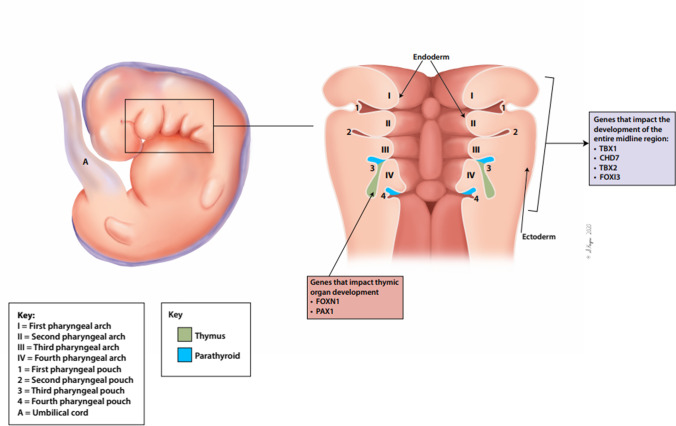
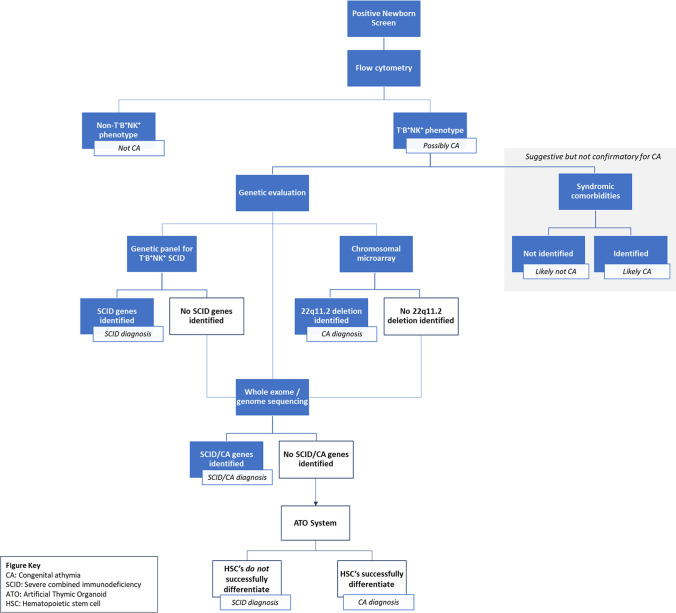
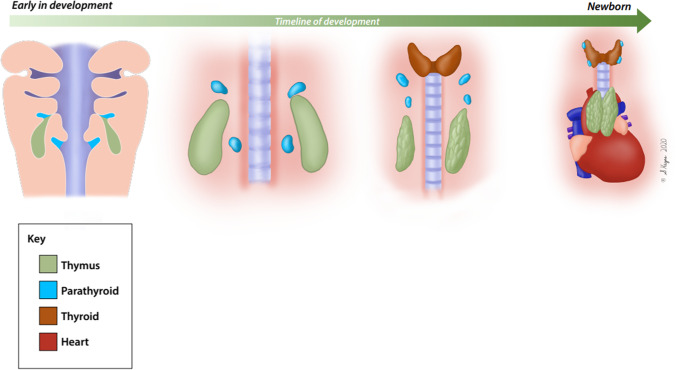
Congenital athymia is an ultra-rare disease characterized by the absence of a functioning thymus. It is associated with several genetic and syndromic disorders including FOXN1 deficiency, 22q11.2 deletion, CHARGE Syndrome (Coloboma, Heart defects, Atresia of the nasal choanae, Retardation of growth and development, Genitourinary anomalies, and Ear anomalies), and Complete DiGeorge Syndrome. Congenital athymia can result from defects in genes that impact thymic organ development such as FOXN1 and PAX1 or from genes that are involved in development of the entire midline region, such as TBX1 within the 22q11.2 region, CHD7, and FOXI3. Patients with congenital athymia have profound immunodeficiency, increased susceptibility to infections, and frequently, autologous graft-versus-host disease (GVHD). Athymic patients often present with absent T cells but normal numbers of B cells and Natural Killer cells (TBNK), similar to a phenotype of severe combined immunodeficiency (SCID); these patients may require additional steps to confirm the diagnosis if no known genetic cause of athymia is identified. However, distinguishing athymia from SCID is crucial, as treatments differ for these conditions. Cultured thymus tissue is being investigated as a treatment for congenital athymia. Here, we review what is known about the epidemiology, underlying etiologies, clinical manifestations, and treatments for congenital athymia.
先天性无胸腺是一种极罕见的疾病,其特征是胸腺功能缺失。它与几种遗传和综合征疾病有关,包括 FOXN1 缺乏症、22q11.2 缺失、 CHARGE 综合征(眼裂畸形、心脏缺陷、鼻后孔闭锁、生长发育迟缓、泌尿生殖系统异常和耳部异常)和完全性 DiGeorge 综合征。先天性无胸腺可能是由于影响胸腺器官发育的基因缺陷引起的,如 FOXN1 和 PAX1,也可能是由于涉及整个中线区域发育的基因缺陷引起的,如 22q11.2 区域内的 TBX1、CHD7 和 FOXI3。先天性无胸腺患者存在严重免疫缺陷,易感染,且常发生自体移植物抗宿主病(GVHD)。无胸腺患者通常表现为 T 细胞缺失,但 B 细胞和自然杀伤细胞(TBNK)数量正常,类似于严重联合免疫缺陷(SCID)的表型;如果未发现无胸腺的已知遗传原因,这些患者可能需要进一步的步骤来确认诊断。然而,区分无胸腺和 SCID 非常重要,因为这两种疾病的治疗方法不同。培养的胸腺组织正被研究用于治疗先天性无胸腺。在这里,我们回顾了先天性无胸腺的流行病学、潜在病因、临床表现和治疗方法。