Shanghai Key Lab of Intelligent Information Processing, Shanghai, China.
School of Computer Science and Technology, Fudan University, Shanghai, China.
BMC Bioinformatics. 2021 May 14;22(1):249. doi: 10.1186/s12859-021-04165-w.
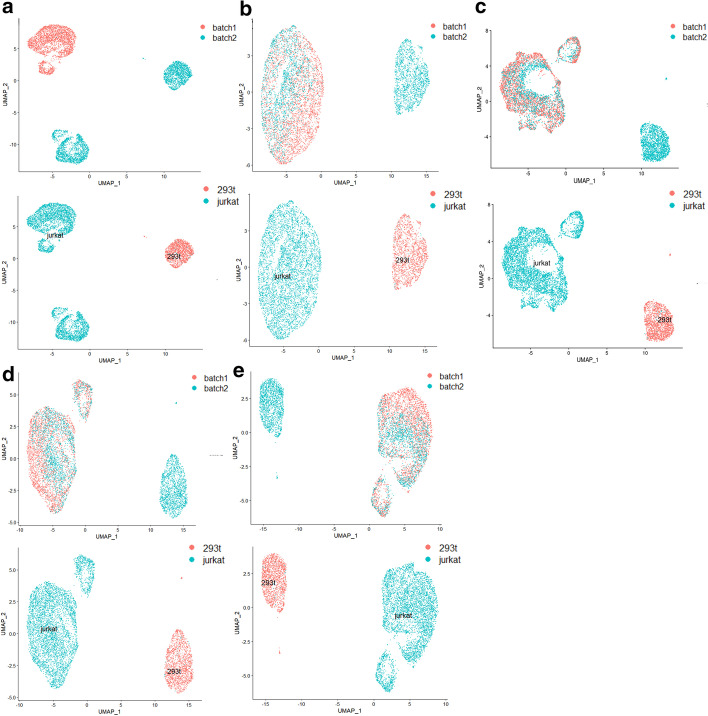
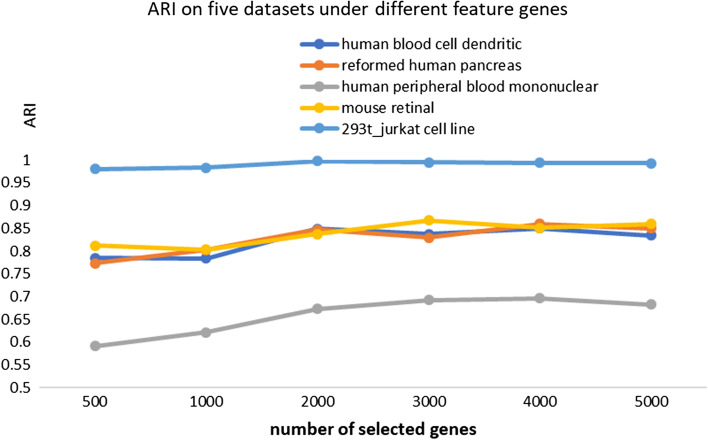
With the continuous maturity of sequencing technology, different laboratories or different sequencing platforms have generated a large amount of single-cell transcriptome sequencing data for the same or different tissues. Due to batch effects and high dimensions of scRNA data, downstream analysis often faces challenges. Although a number of algorithms and tools have been proposed for removing batch effects, the current mainstream algorithms have faced the problem of data overcorrection when the cell type composition varies greatly between batches.
In this paper, we propose a novel method named SSBER by utilizing biological prior knowledge to guide the correction, aiming to solve the problem of poor batch-effect correction when the cell type composition differs greatly between batches.
SSBER effectively solves the above problems and outperforms other algorithms when the cell type structure among batches or distribution of cell population varies considerably, or some similar cell types exist across batches.
随着测序技术的不断成熟,不同的实验室或不同的测序平台针对同一或不同组织产生了大量的单细胞转录组测序数据。由于批次效应和 scRNA 数据的高维性,下游分析通常面临挑战。尽管已经提出了许多用于消除批次效应的算法和工具,但当前主流算法在批次间细胞类型组成差异较大时面临数据过度校正的问题。
本文提出了一种新的方法 SSBER,通过利用生物学先验知识来指导校正,旨在解决批次间细胞类型组成差异较大时批次效应校正效果不佳的问题。
SSBER 有效地解决了上述问题,在批次间细胞类型结构或细胞群体分布差异较大,或存在相似细胞类型的情况下,优于其他算法。