Modi Bhavi P, Khan Haq Nawaz, van der Lee Robin, Wasim Muhammad, Haaxma Charlotte A, Richmond Phillip A, Drögemöller Britt, Shah Suleman, Salomons Gajja, van der Kloet Frans M, Vaz Fred M, van der Crabben Saskia N, Ross Colin J, Wasserman Wyeth W, van Karnebeek Clara D M, Awan Fazli Rabbi
Centre for Molecular Medicine and Therapeutics, Dept. of Medical Genetics, BC Children's Hospital Research Institute, University of British Columbia, Vancouver, BC, Canada.
Health Biotechnology Division, National Institute for Biotechnology and Genetic Engineering (NIBGE), Faisalabad, Pakistan.
Mol Genet Metab Rep. 2021 Apr 26;27:100761. doi: 10.1016/j.ymgmr.2021.100761. eCollection 2021 Jun.
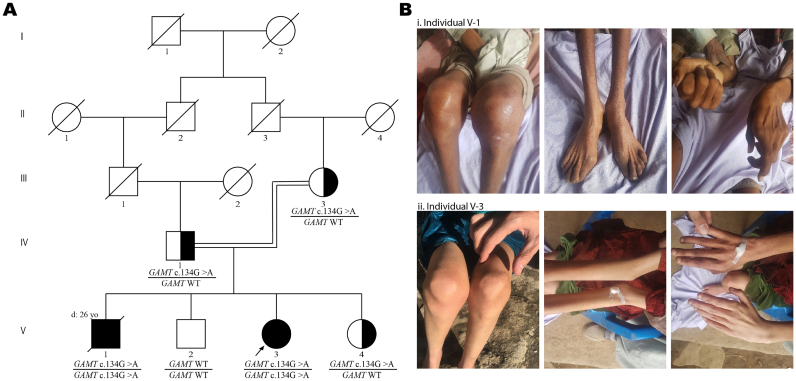
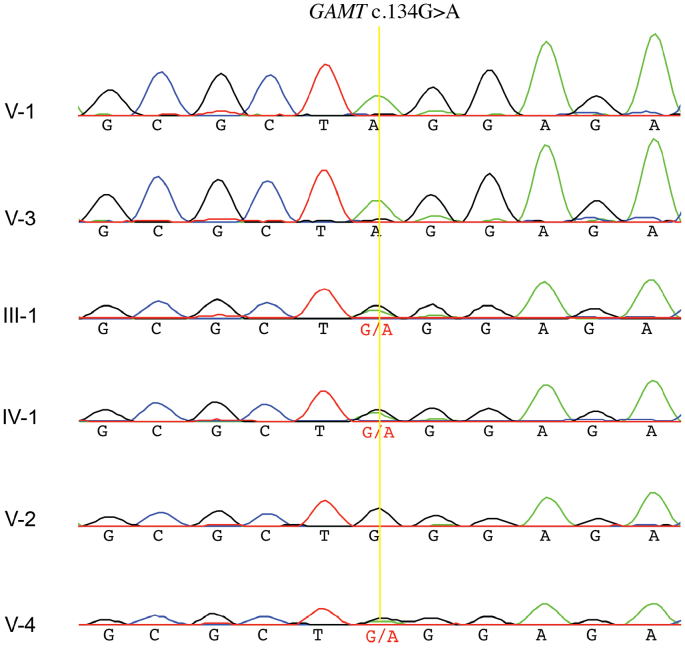
Guanidinoacetate methyltransferase (GAMT) deficiency is a creatine deficiency disorder and an inborn error of metabolism presenting with progressive intellectual and neurological deterioration. As most cases are identified and treated in early childhood, adult phenotypes that can help in understanding the natural history of the disorder are rare. We describe two adult cases of GAMT deficiency from a consanguineous family in Pakistan that presented with a history of global developmental delay, cognitive impairments, excessive drooling, behavioral abnormalities, contractures and apparent bone deformities initially presumed to be the reason for abnormal gait. Exome sequencing identified a homozygous nonsense variant in : NM_000156.5:c.134G>A (p.Trp45*). We also performed a literature review and compiled the genetic and clinical characteristics of all adult cases of GAMT deficiency reported to date. When compared to the adult cases previously reported, the musculoskeletal phenotype and the rapidly progressive nature of neurological and motor decline seen in our patients is striking. This study presents an opportunity to gain insights into the adult presentation of GAMT deficiency and highlights the need for in-depth evaluation and reporting of clinical features to expand our understanding of the phenotypic spectrum.
胍乙酸甲基转移酶(GAMT)缺乏症是一种肌酸缺乏症,属于先天性代谢缺陷,表现为进行性智力和神经功能衰退。由于大多数病例在儿童早期被确诊和治疗,有助于了解该疾病自然史的成人表型较为罕见。我们描述了来自巴基斯坦一个近亲家庭的两例成人GAMT缺乏症病例,他们有全球发育迟缓、认知障碍、流涎过多、行为异常、挛缩和明显骨骼畸形的病史,最初认为这些是异常步态的原因。外显子组测序在NM_000156.5:c.134G>A(p.Trp45*)中鉴定出一个纯合无义变异。我们还进行了文献综述,并汇总了迄今为止报道的所有成人GAMT缺乏症病例的遗传和临床特征。与先前报道的成人病例相比,我们患者中出现的肌肉骨骼表型以及神经和运动功能衰退的快速进展性质令人瞩目。本研究为深入了解成人GAMT缺乏症的表现提供了契机,并强调了深入评估和报告临床特征以扩大我们对表型谱理解的必要性。