Pedrood Keyvan, Azizian Homa, Montazer Mohammad Nazari, Mohammadi-Khanaposhtani Maryam, Asgari Mohammad Sadegh, Asadi Mehdi, Bahadorikhalili Saeed, Rastegar Hossein, Larijani Bagher, Amanlou Massoud, Mahdavi Mohammad
Endocrinology and Metabolism Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran.
Department of Medicinal Chemistry, School of Pharmacy, Iran University of Medical Sciences, Tehran, Iran.
Sci Rep. 2021 May 19;11(1):10607. doi: 10.1038/s41598-021-90104-x.
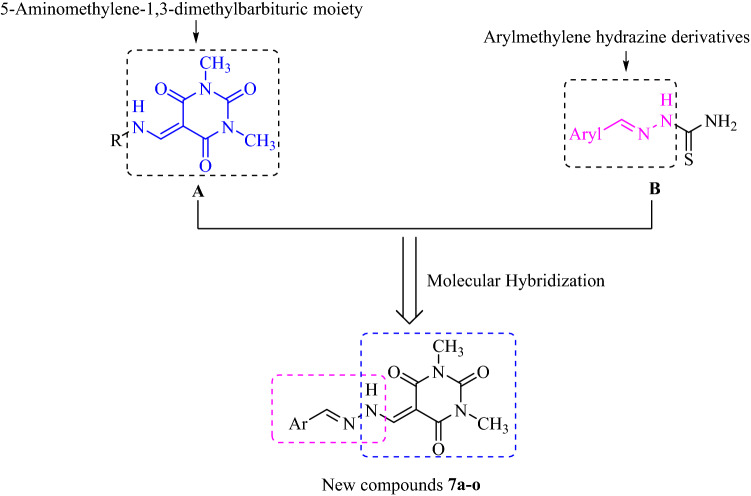
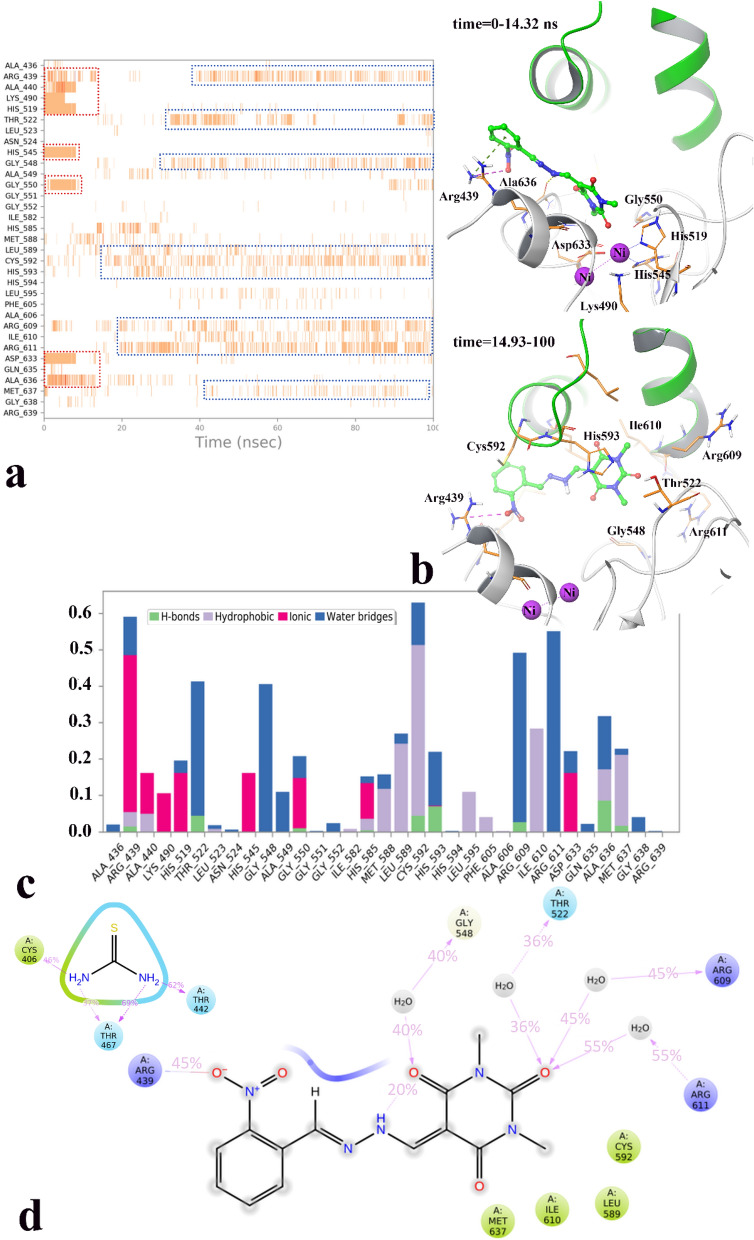
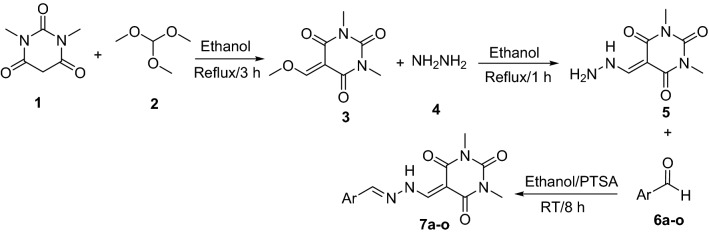
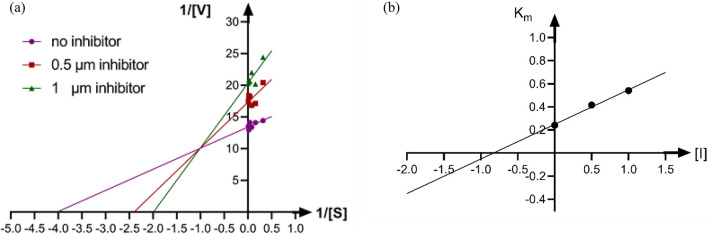
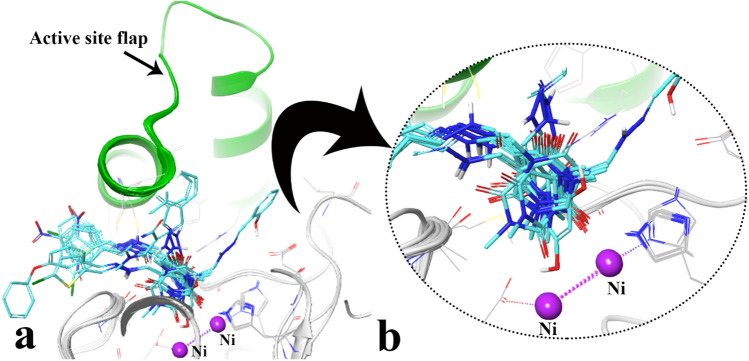
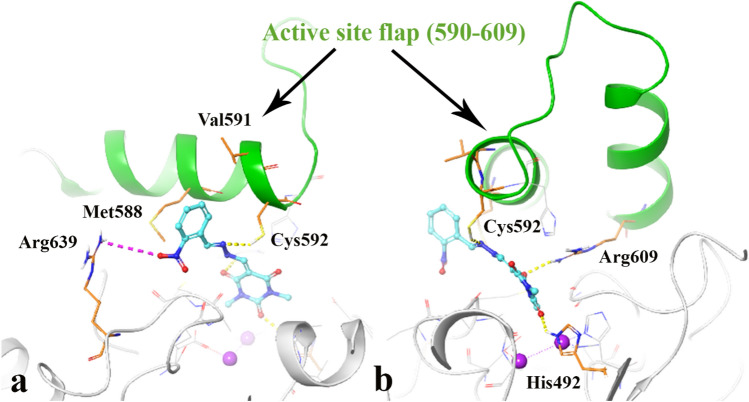
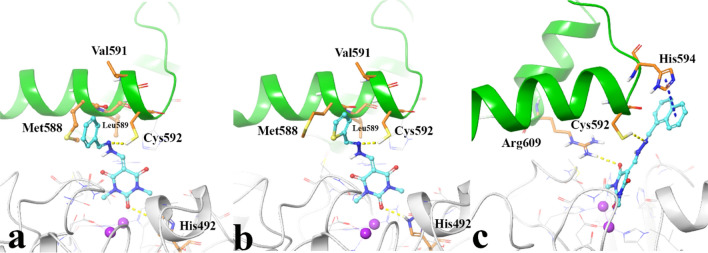
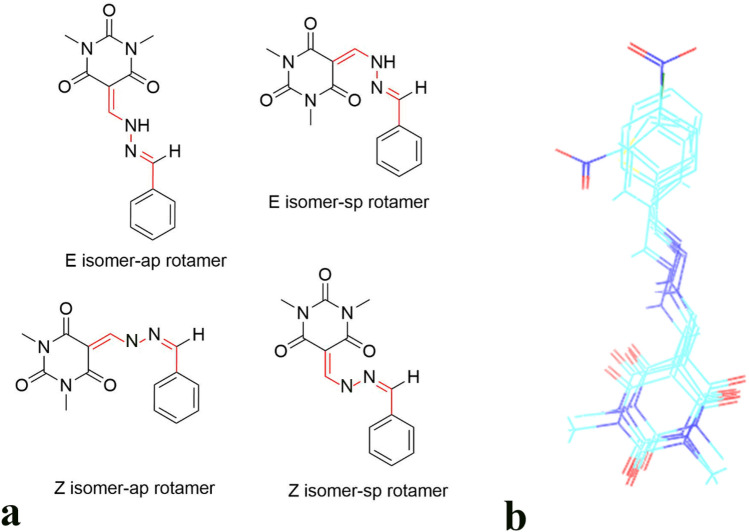
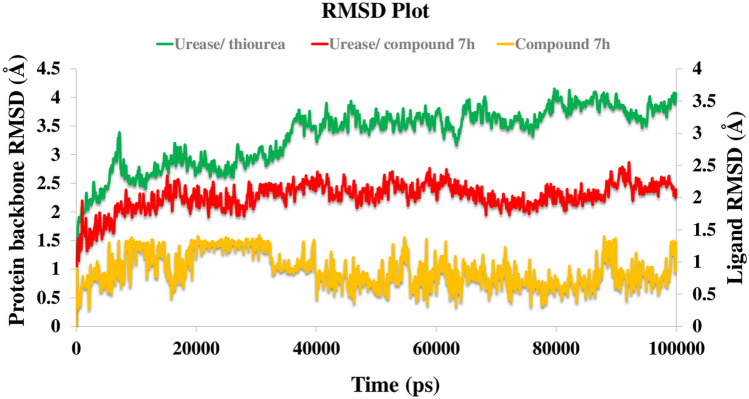
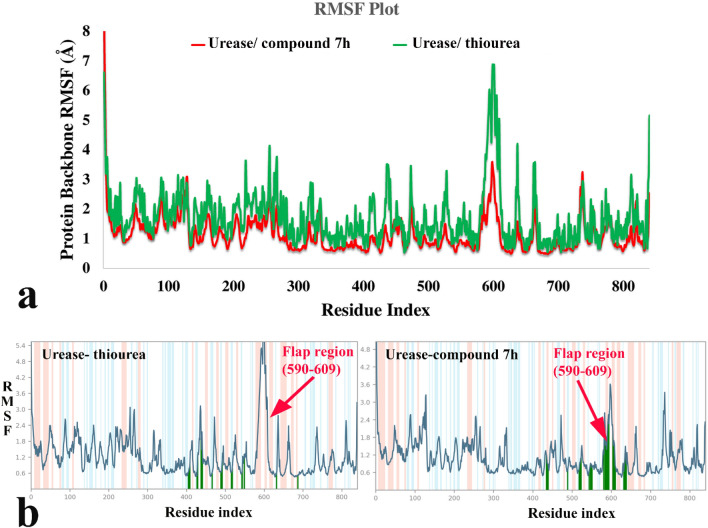
A new series of arylmethylene hydrazine derivatives bearing 1,3-dimethylbarbituric moiety 7a-o were designed, synthesized, and evaluated for their in vitro urease inhibitory activity. All the title compounds displayed high anti-urease activity, with IC values in the range of 0.61 ± 0.06-4.56 ± 0.18 µM as compared to the two standard inhibitors hydroxyurea (IC = 100 ± 0.15 μM) and thiourea (IC = 23 ± 1.7 μM). Among the synthesized compounds, compound 7h with 2-nitro benzylidene group was found to be the most potent compound. Kinetic study of this compound revealed that it is a mix-mode inhibitor against urease. Evaluation of the interaction modes of the synthesized compounds in urease active site by molecular modeling revealed that that compounds with higher urease inhibitor activity (7h, 7m, 7c, 7l, 7i, and 7o, with IC of 0.61, 0.86, 1.2, 1.34, 1.33, 1.94 μM, respectively) could interact with higher number of residues, specially Arg609, Cys592 (as part of urease active site flap) and showed higher computed free energy, while compounds with lower urease activity (7f, 7n, 7g, and 7a with IC of 3.56, 4.56, 3.62 and 4.43 μM, respectively) and could not provide the proper interaction with Arg609, and Cys592 as the key interacting residues along with lower free binding energy. MD investigation revealed compound 7h interacted with Arg609 and Cys592 which are of the key residues at the root part of mobile flap covering the active site. Interacting with the mentioned residue for a significant amount of time, affects the flexibility of the mobile flap covering the active site and causes inhibition of the ureolytic activity. Furthermore, in silico physico-chemical study of compounds 7a-o predicted that all these compounds are drug-likeness with considerable orally availability.
设计、合成了一系列带有1,3 - 二甲基巴比妥部分的新型芳基亚甲基肼衍生物7a - o,并对其体外脲酶抑制活性进行了评估。与两种标准抑制剂羟基脲(IC = 100±0.15 μM)和硫脲(IC = 23±1.7 μM)相比,所有目标化合物均表现出高抗脲酶活性,IC值在0.61±0.06 - 4.56±0.18 μM范围内。在合成的化合物中,带有2 - 硝基苄叉基的化合物7h被发现是最有效的化合物。该化合物的动力学研究表明它是一种针对脲酶的混合模式抑制剂。通过分子建模评估合成化合物在脲酶活性位点的相互作用模式表明,脲酶抑制活性较高的化合物(7h、7m、7c、7l、7i和7o,IC分别为0.61、0.86、1.2、1.34、1.33、1.94 μM)可以与更多数量的残基相互作用,特别是Arg609、Cys592(作为脲酶活性位点瓣的一部分),并显示出更高的计算自由能,而脲酶活性较低的化合物(7f、7n、7g和7a,IC分别为3.56、4.56、3.62和4.43 μM)不能与Arg609和Cys592这两个关键相互作用残基进行适当的相互作用,并且结合自由能较低。分子动力学研究表明化合物7h与Arg609和Cys592相互作用,这两个残基是覆盖活性位点的可移动瓣根部的关键残基。与上述残基长时间相互作用会影响覆盖活性位点的可移动瓣的灵活性,并导致脲解活性受到抑制。此外,对化合物7a - o的计算机辅助物理化学研究预测,所有这些化合物都具有药物相似性,口服可用性相当高。