Endocrinology and Metabolism Research Center, Endocrinology and Metabolism Clinical Sciences Institute, Tehran University of Medical Sciences, Tehran, Iran.
Department of Medicinal Chemistry, School of Pharmacy, Iran University of Medical Sciences, Tehran, Iran.
Sci Rep. 2022 Aug 15;12(1):13827. doi: 10.1038/s41598-022-17993-4.
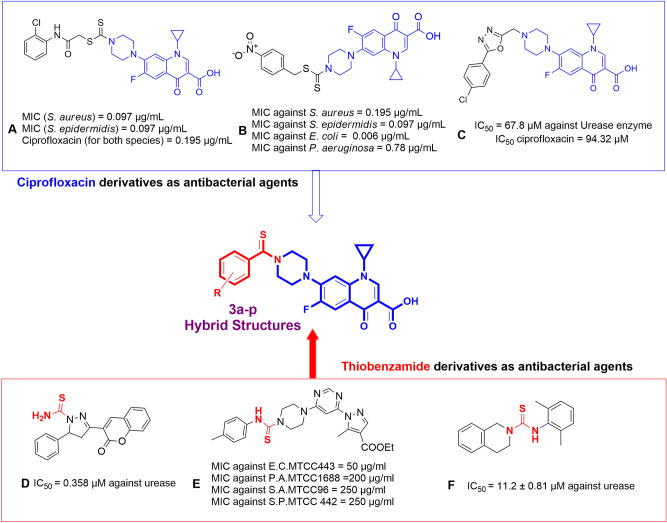
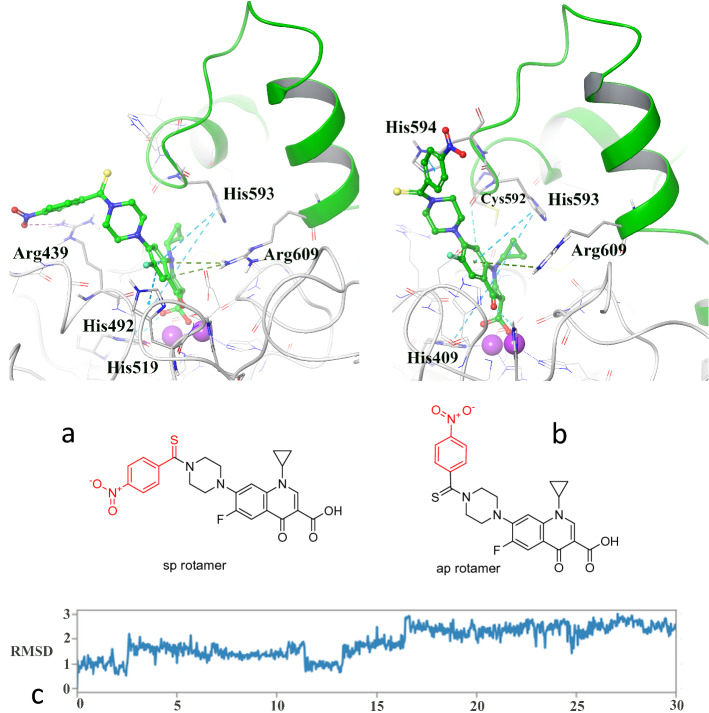
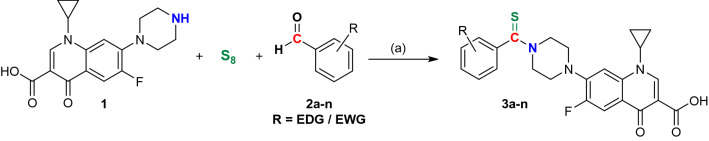
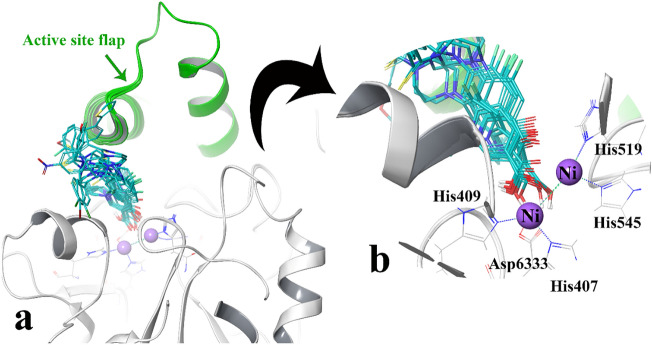
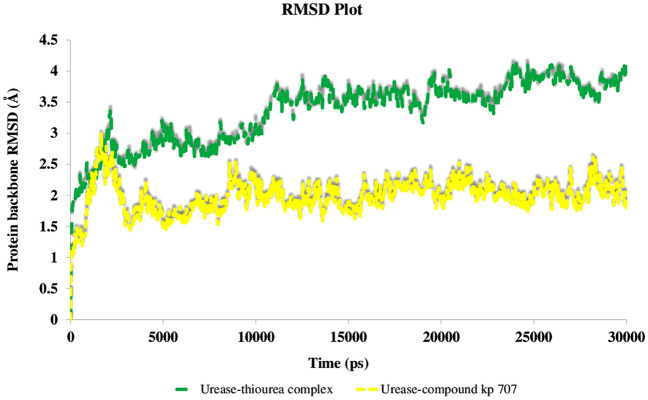
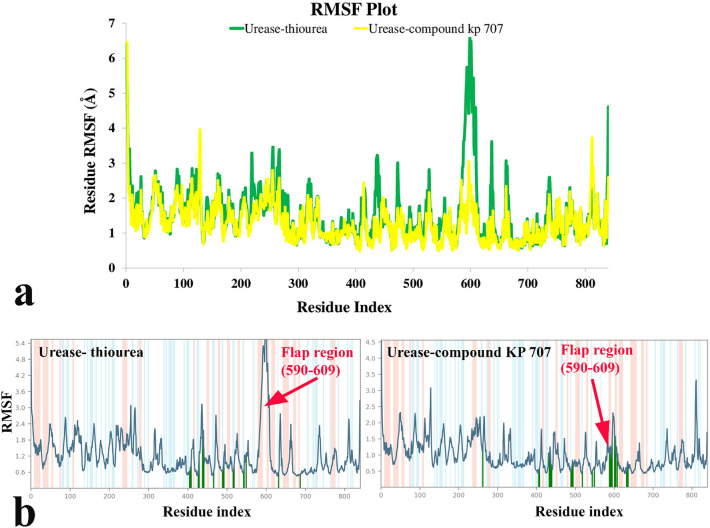
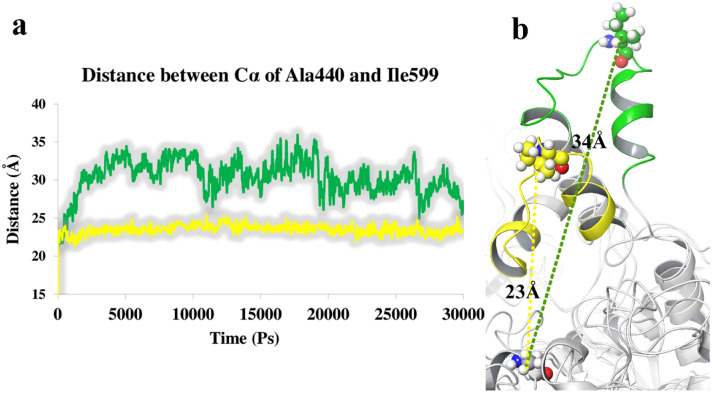
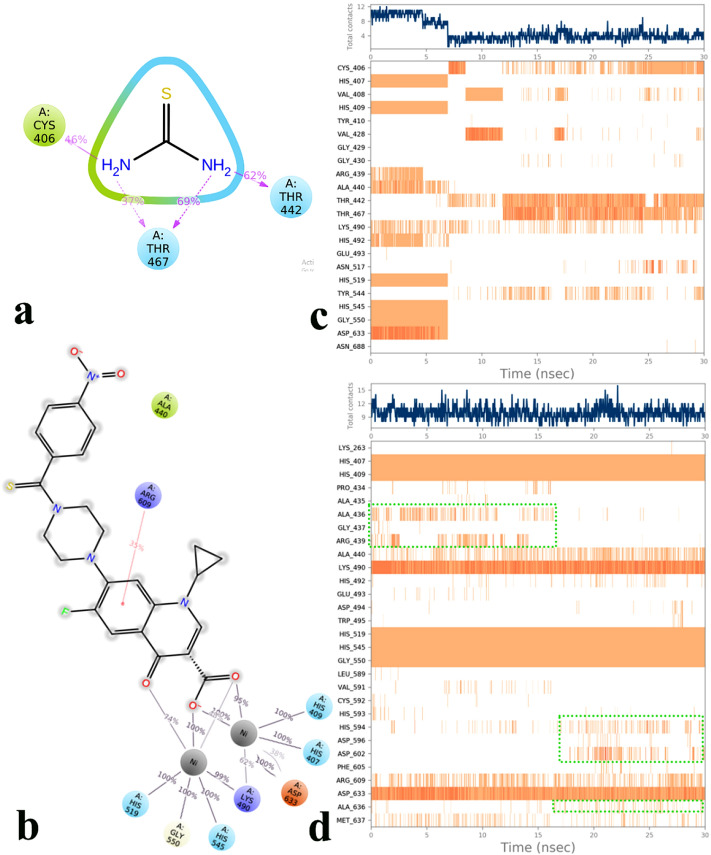
A new series of N-thioacylated ciprofloxacin 3a-n were designed and synthesized based on Willgerodt-Kindler reaction. The results of in vitro urease inhibitory assay indicated that almost all the synthesized compounds 3a-n (IC = 2.05 ± 0.03-32.49 ± 0.32 μM) were more potent than standard inhibitors, hydroxyurea (IC = 100 ± 2.5 μM) and thiourea (IC = 23 ± 0.84 μM). The study of antibacterial activity against Gram-positive species (S. aureus and S. epidermidis) revealed that the majority of compounds were more active than ciprofloxacin as the standard drug, and 3h derivative bearing 3-fluoro group had the same effect as ciprofloxacin against Gram-negative bacteria (P. aeruginosa and E. coli). Based on molecular dynamic simulations, compound 3n exhibited pronounced interactions with the critical residues of the urease active site and mobile flap pocket so that the quinolone ring coordinated toward the metal bi-nickel center and the essential residues at the flap site like His593, His594, and Arg609. These interactions caused blocking the active site and stabilized the movement of the mobile flap at the entrance of the active site channel, which significantly reduced the catalytic activity of urease. Noteworthy, 3n also exhibited IC values of 5.59 ± 2.38 and 5.72 ± 1.312 µg/ml to inhibit urease enzyme against C. neoformans and P. vulgaris in the ureolytic assay.
基于Willgerodt-Kindler 反应,设计并合成了一系列新的 N-硫代乙酰化环丙沙星 3a-n。体外脲酶抑制实验结果表明,几乎所有合成的化合物 3a-n(IC50=2.05±0.03-32.49±0.32 μM)均比标准抑制剂羟基脲(IC50=100±2.5 μM)和硫脲(IC50=23±0.84 μM)具有更强的抑制活性。对革兰氏阳性菌(金黄色葡萄球菌和表皮葡萄球菌)的抗菌活性研究表明,大多数化合物比作为标准药物的环丙沙星更具活性,而带有 3-氟取代基的 3h 衍生物对革兰氏阴性菌(铜绿假单胞菌和大肠杆菌)的活性与环丙沙星相同。基于分子动力学模拟,化合物 3n 与脲酶活性位点的关键残基和可移动瓣口袋表现出明显的相互作用,使得喹诺酮环与金属双镍中心以及瓣位的必需残基(His593、His594 和 Arg609)配位。这些相互作用导致了活性位点的阻断,并稳定了活性位点通道入口处可移动瓣的运动,从而显著降低了脲酶的催化活性。值得注意的是,3n 在脲酶抑制试验中对新生隐球菌和普通变形杆菌的脲酶抑制 IC 值分别为 5.59±2.38 和 5.72±1.312 μg/ml。