Zhang Tongxia, Yan Chuanzhu, Liu Yiming, Cao Lili, Ji Kunqian, Li Duoling, Chi Lingyi, Zhao Yuying
Research Institute of Neuromuscular and Neurodegenerative Diseases and Department of Neurology, Qilu Hospital, Shandong University, Jinan, People's Republic of China.
School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, People's Republic of China.
Neuropsychiatr Dis Treat. 2021 May 12;17:1451-1458. doi: 10.2147/NDT.S296424. eCollection 2021.
Leukodystrophies are frequently regarded as childhood disorders, but they can occur at any age, and the clinical and imaging patterns of the adult-onset form are usually different from the better-known childhood variants. Several reports have shown that various late-onset leukodystrophies, such as X-linked adrenoleukodystrophy and Krabbe disease, may present as spastic paraplegia with the absence of the characteristic white matter lesions on neuroimaging; this can be easily misdiagnosed as hereditary spastic paraplegia. The objective of this study was to investigate the frequency of late-onset leukodystrophies in patients with spastic paraplegia.
We performed genetic analysis using a custom-designed gene panel for leukodystrophies in 112 hereditary spastic paraplegia-like patients.
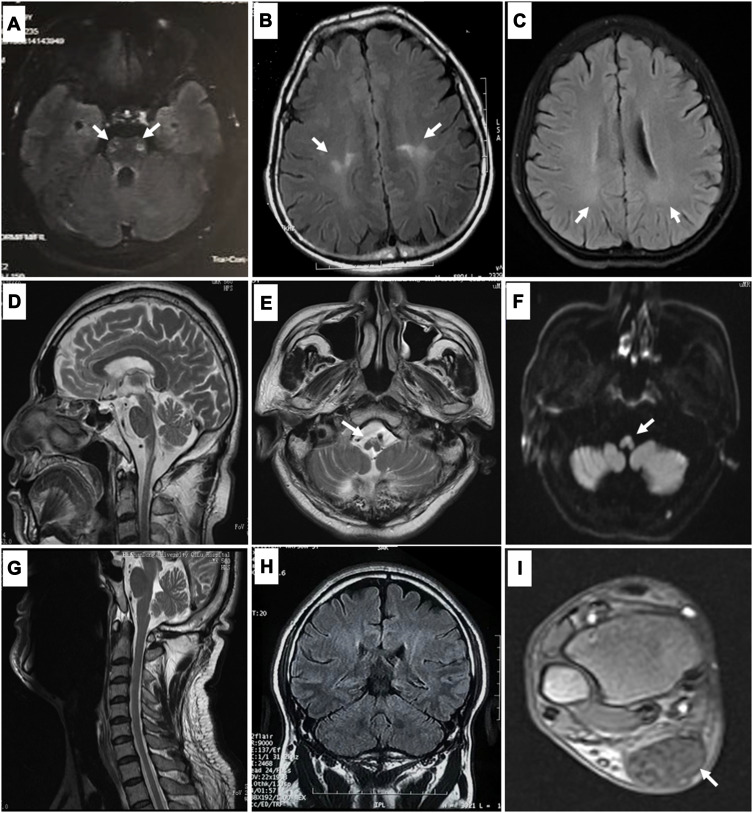
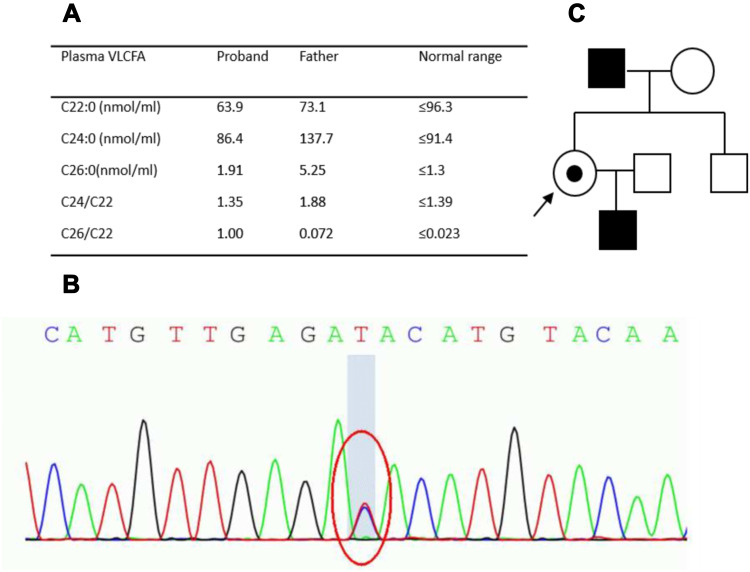
We identified pathogenic mutations in 13 out of 112 patients, including five patients with adrenomyeloneuropathy, three with Krabbe disease, three with Alexander disease, and two with cerebrotendinous xanthomatosis. In terms of clinical manifestations, in addition to spastic paraplegia, three adrenomyeloneuropathy probands also had adrenocortical insufficiency, two Alexander disease probands developed urinary retention, one CTX proband developed cataracts and chronic diarrhea and the other presented with chronic diarrhea and mild tendon xanthomatosis. None of the patients had evidence of diffuse leukodystrophy on neuroimaging.
Patients with late-onset spastic paraplegia should be screened for underlying leukodystrophies, irrespective of the presence of additional complicating symptoms and neuroimaging abnormalities.
脑白质营养不良通常被视为儿童疾病,但可发生于任何年龄,成人起病型的临床和影像学表现通常与更为人熟知的儿童型不同。多项报告显示,多种迟发性脑白质营养不良,如X连锁肾上腺脑白质营养不良和克拉伯病,可能表现为痉挛性截瘫,神经影像学上无特征性白质病变;这很容易被误诊为遗传性痉挛性截瘫。本研究的目的是调查痉挛性截瘫患者中迟发性脑白质营养不良的发生率。
我们对112例遗传性痉挛性截瘫样患者使用定制设计的脑白质营养不良基因检测板进行了基因分析。
我们在112例患者中鉴定出13例致病突变,其中5例患有肾上腺脊髓神经病,3例患有克拉伯病,3例患有亚历山大病,2例患有脑腱黄瘤病。在临床表现方面,除痉挛性截瘫外,3例肾上腺脊髓神经病先证者还患有肾上腺皮质功能不全,2例亚历山大病先证者出现尿潴留,1例脑腱黄瘤病先证者出现白内障和慢性腹泻,另1例表现为慢性腹泻和轻度肌腱黄瘤。所有患者神经影像学均无弥漫性脑白质营养不良的证据。
迟发性痉挛性截瘫患者应筛查潜在的脑白质营养不良,无论是否存在其他并发症状和神经影像学异常。