Malakar Tanmay, Hanson Carly S, Devery James J, Zimmerman Paul M
Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, United States.
Department of Chemistry & Biochemistry, Loyola University Chicago, Chicago, Illinois 60660, United States.
ACS Catal. 2021 Apr 16;11(8):4381-4394. doi: 10.1021/acscatal.0c05277. Epub 2021 Mar 25.
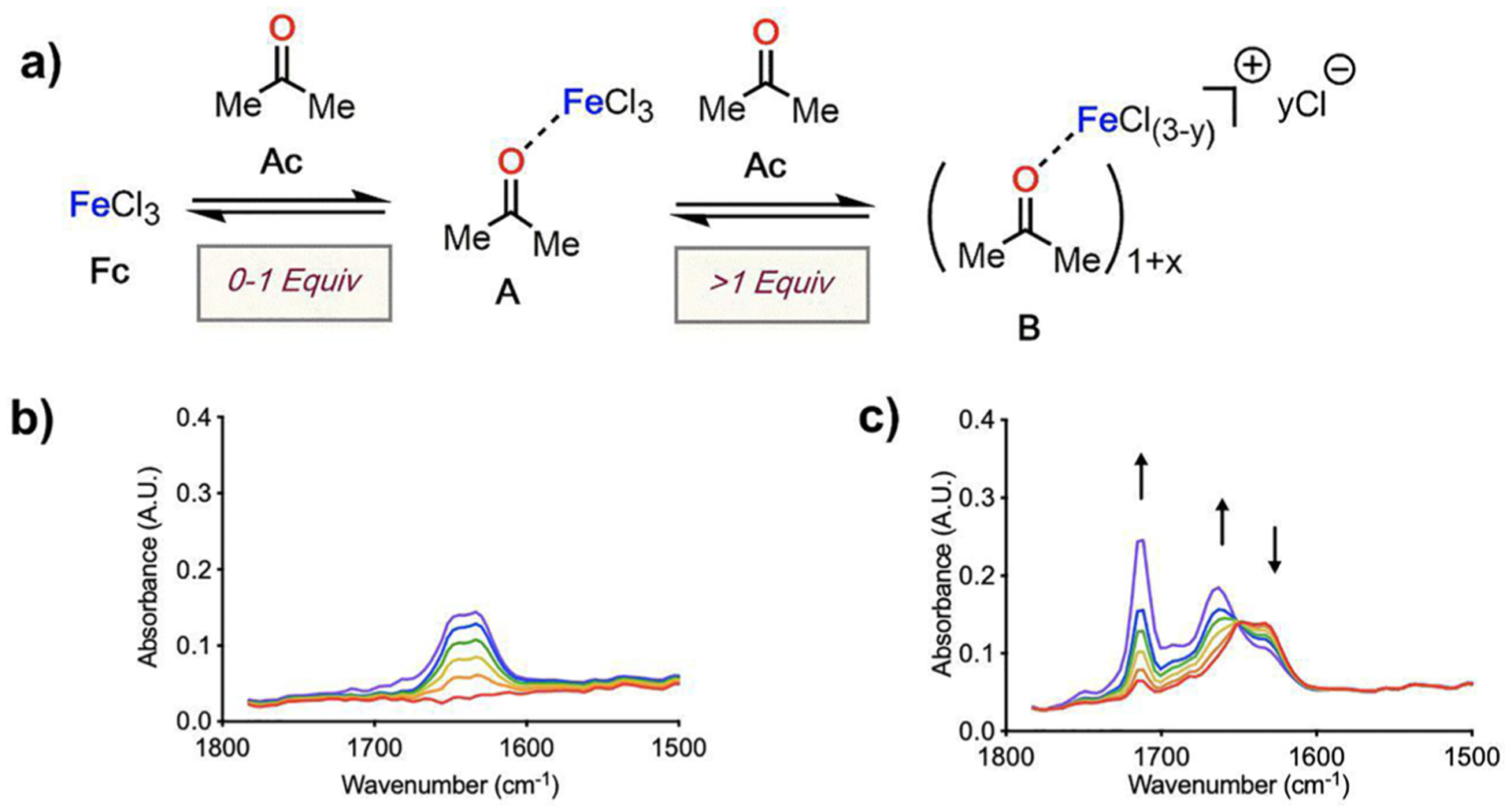
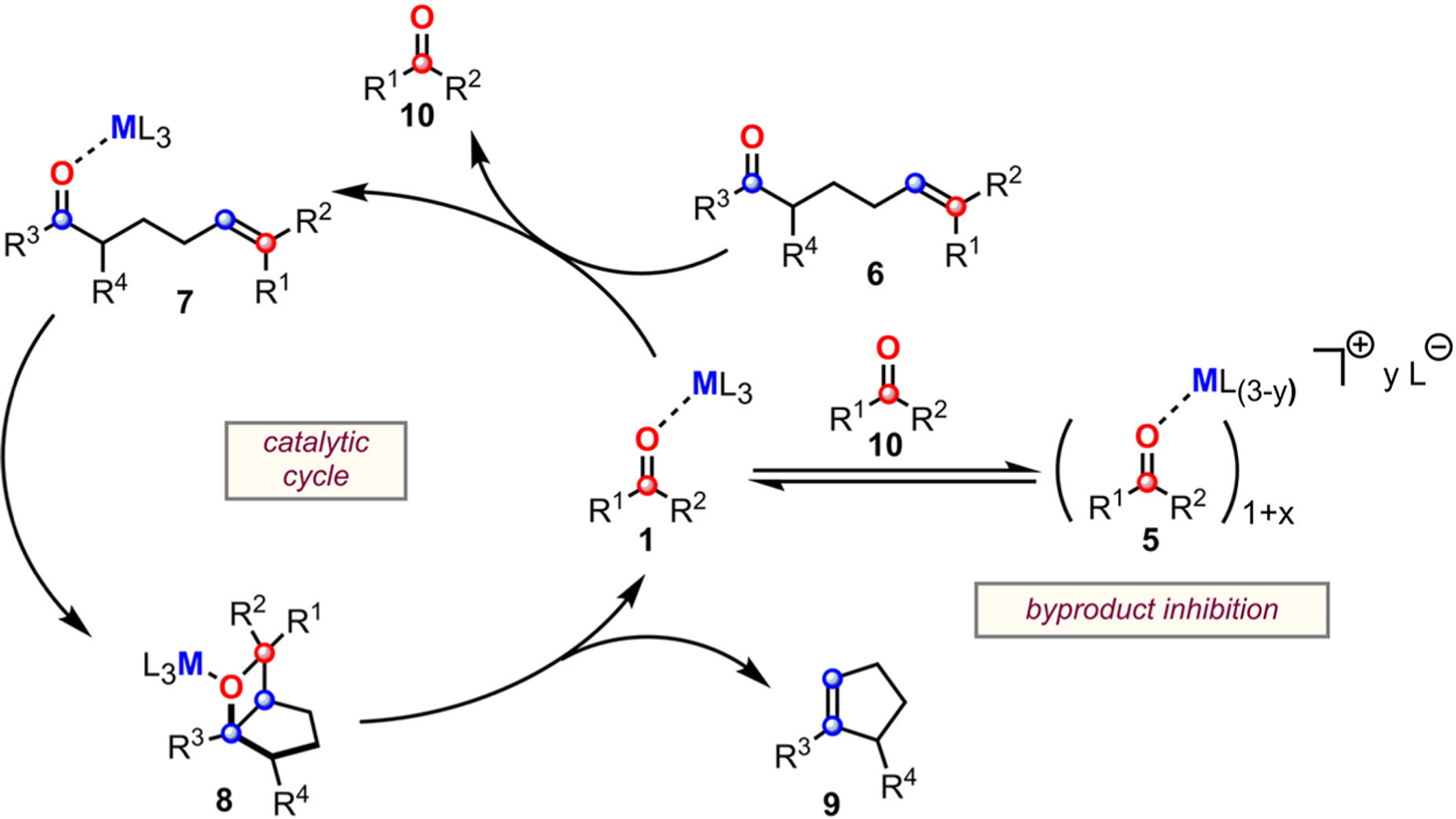
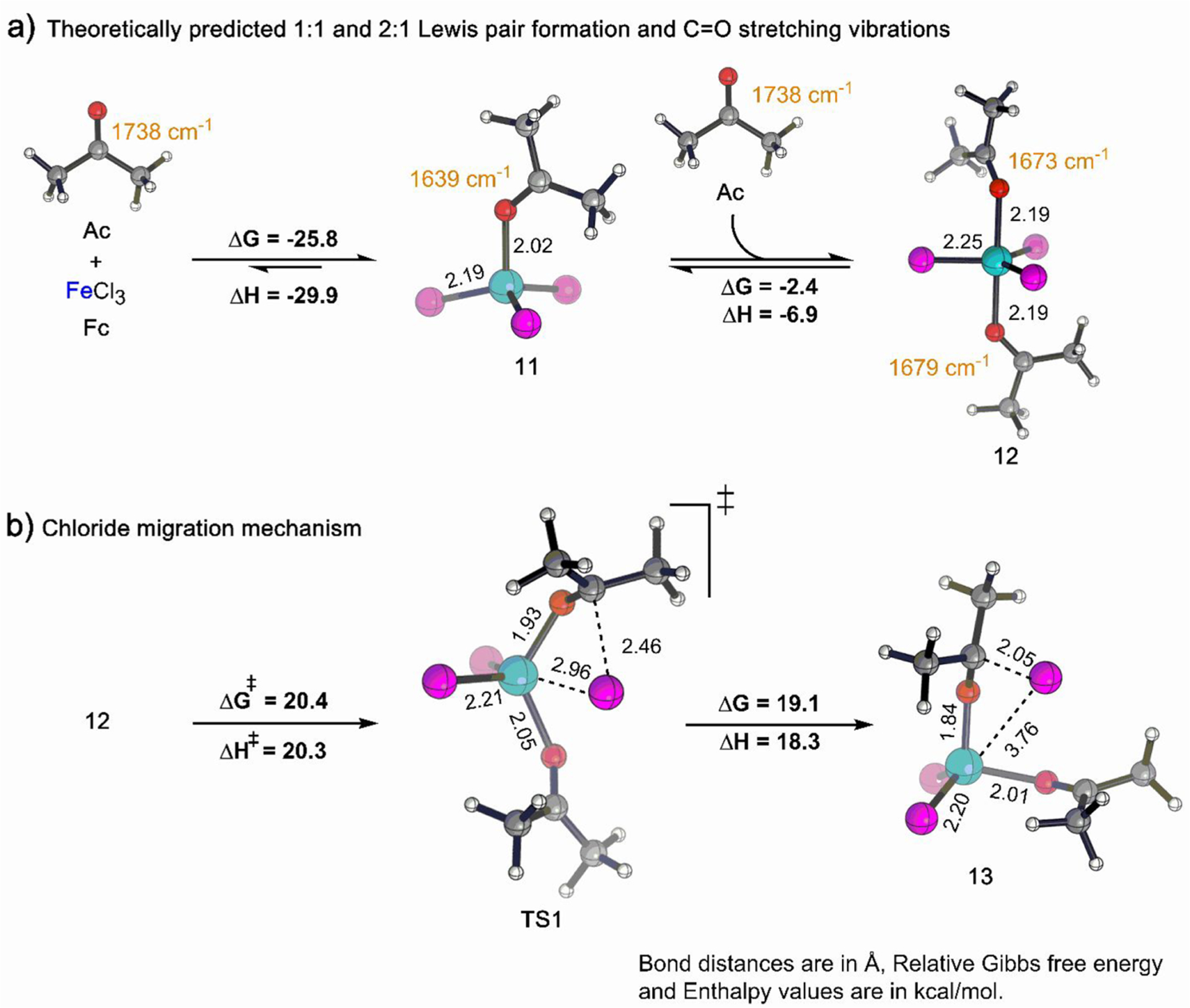
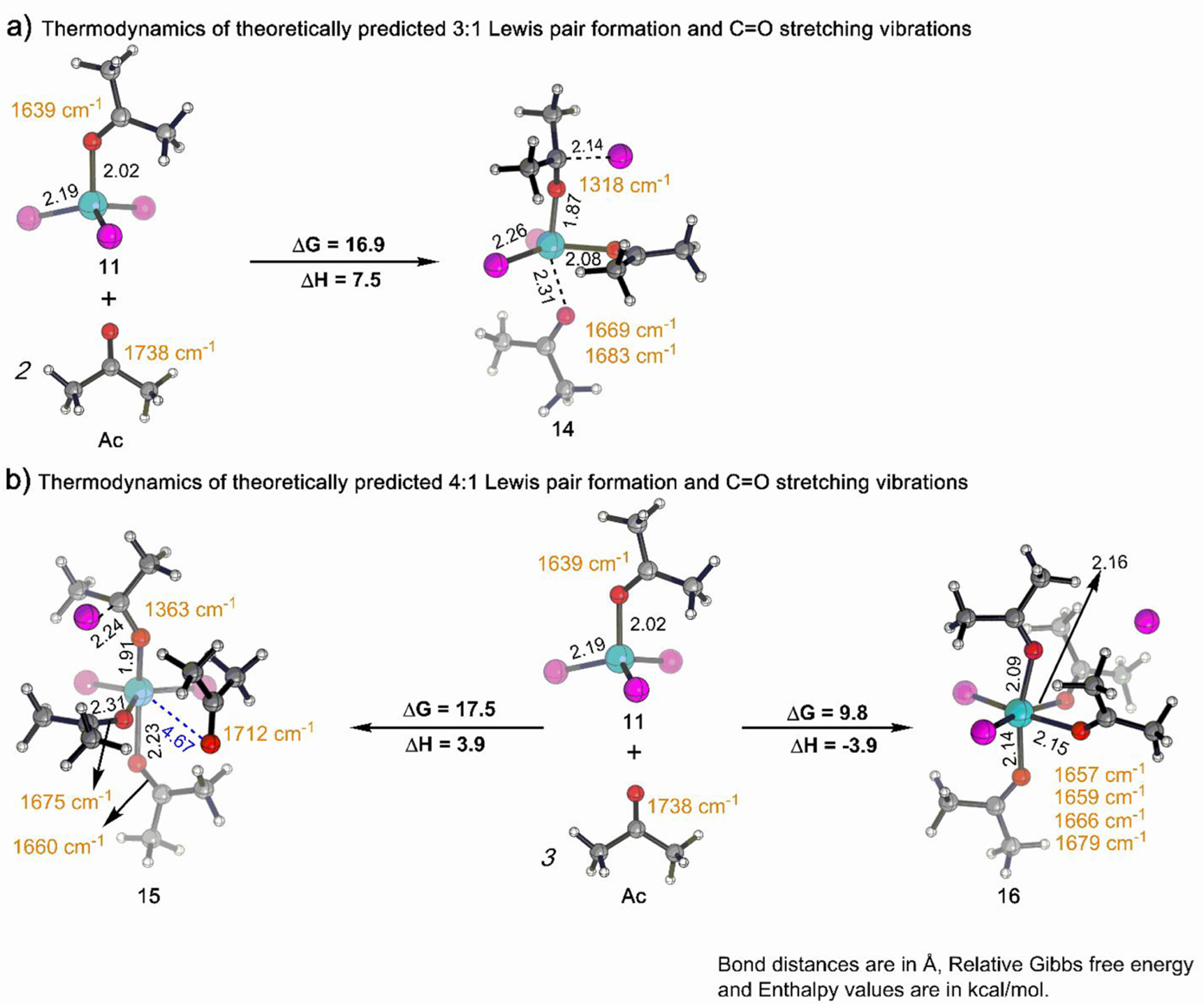
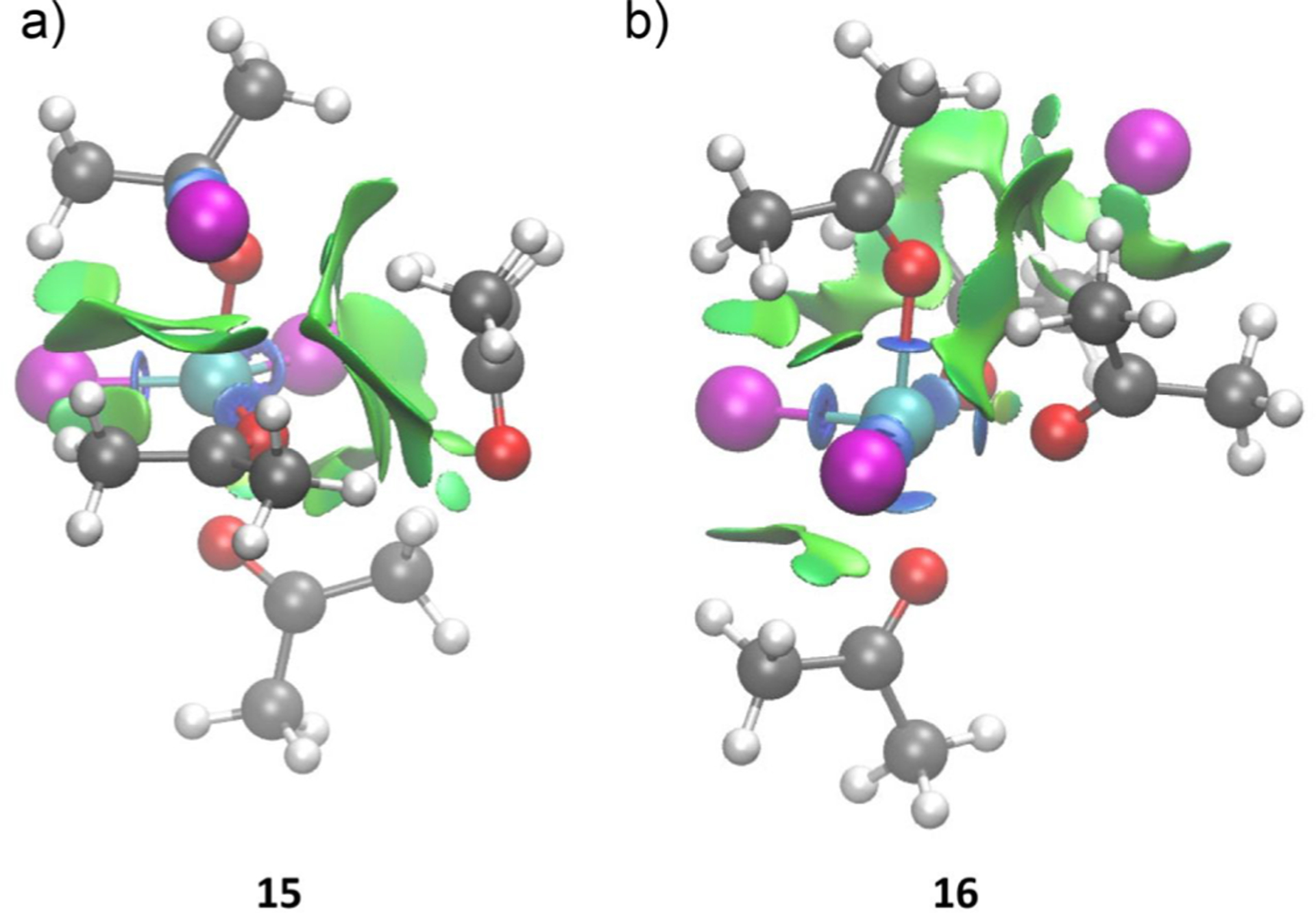
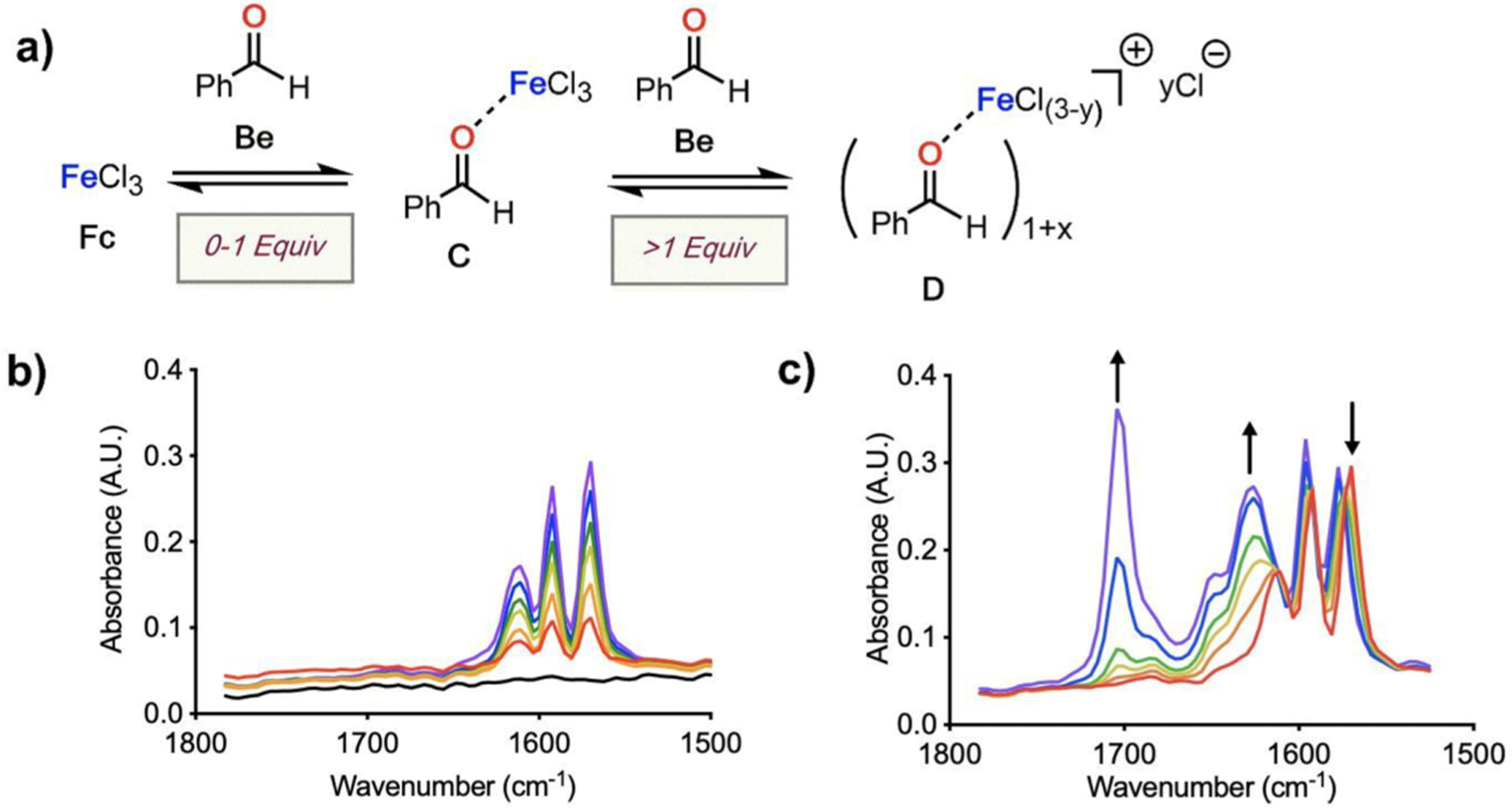
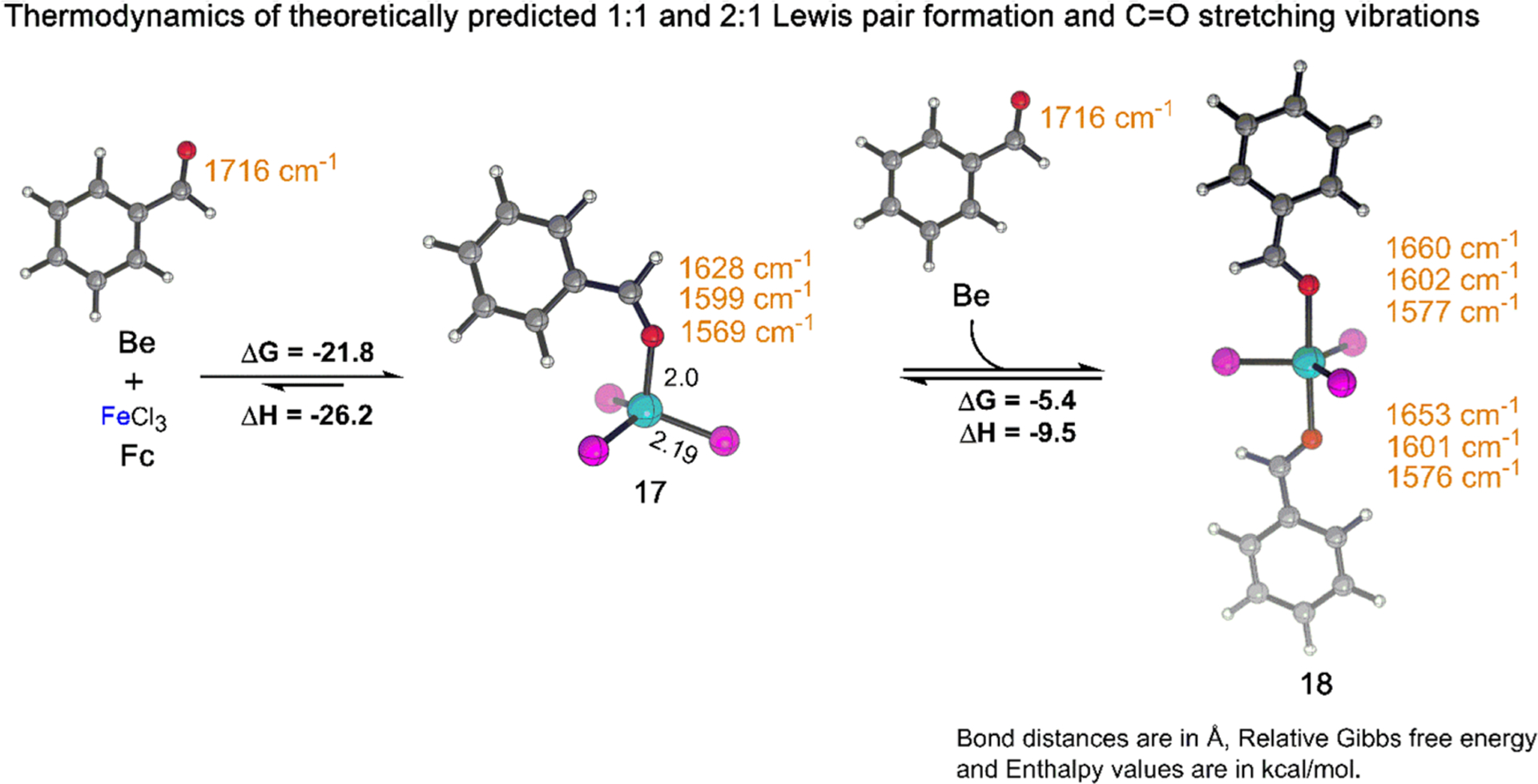
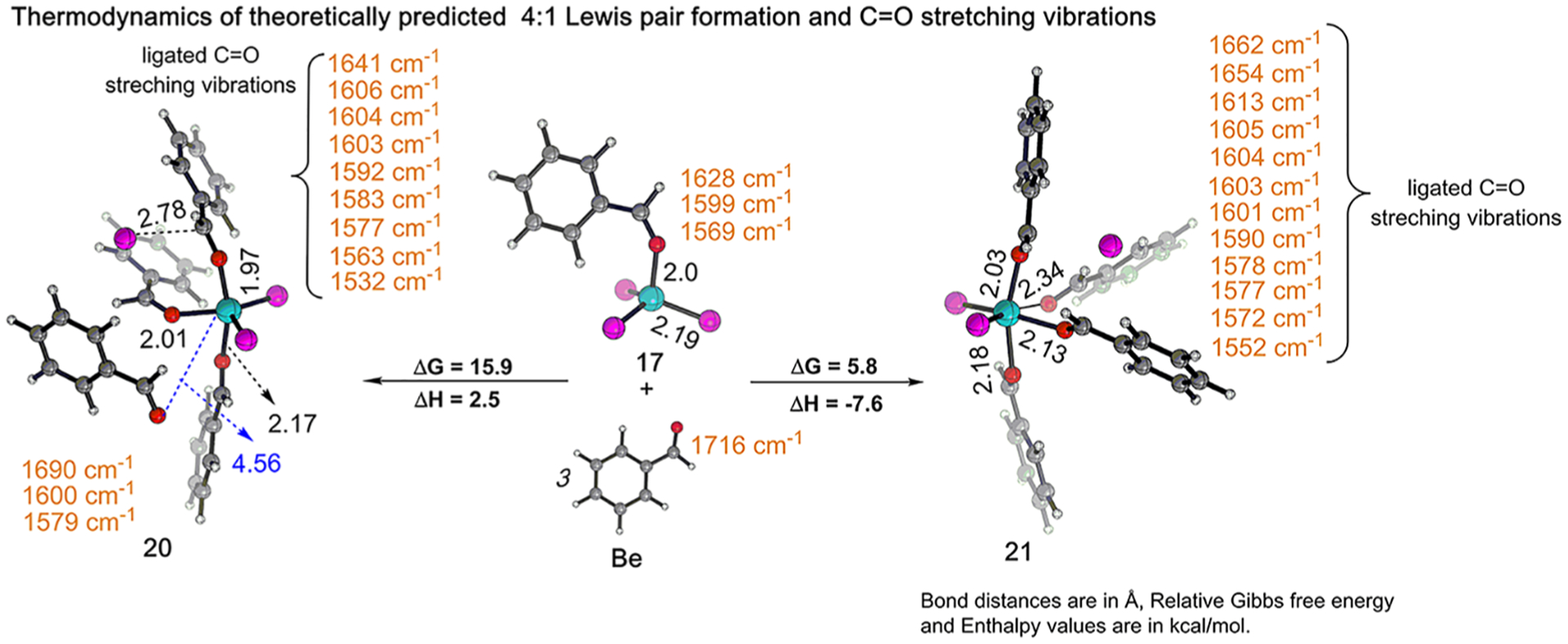
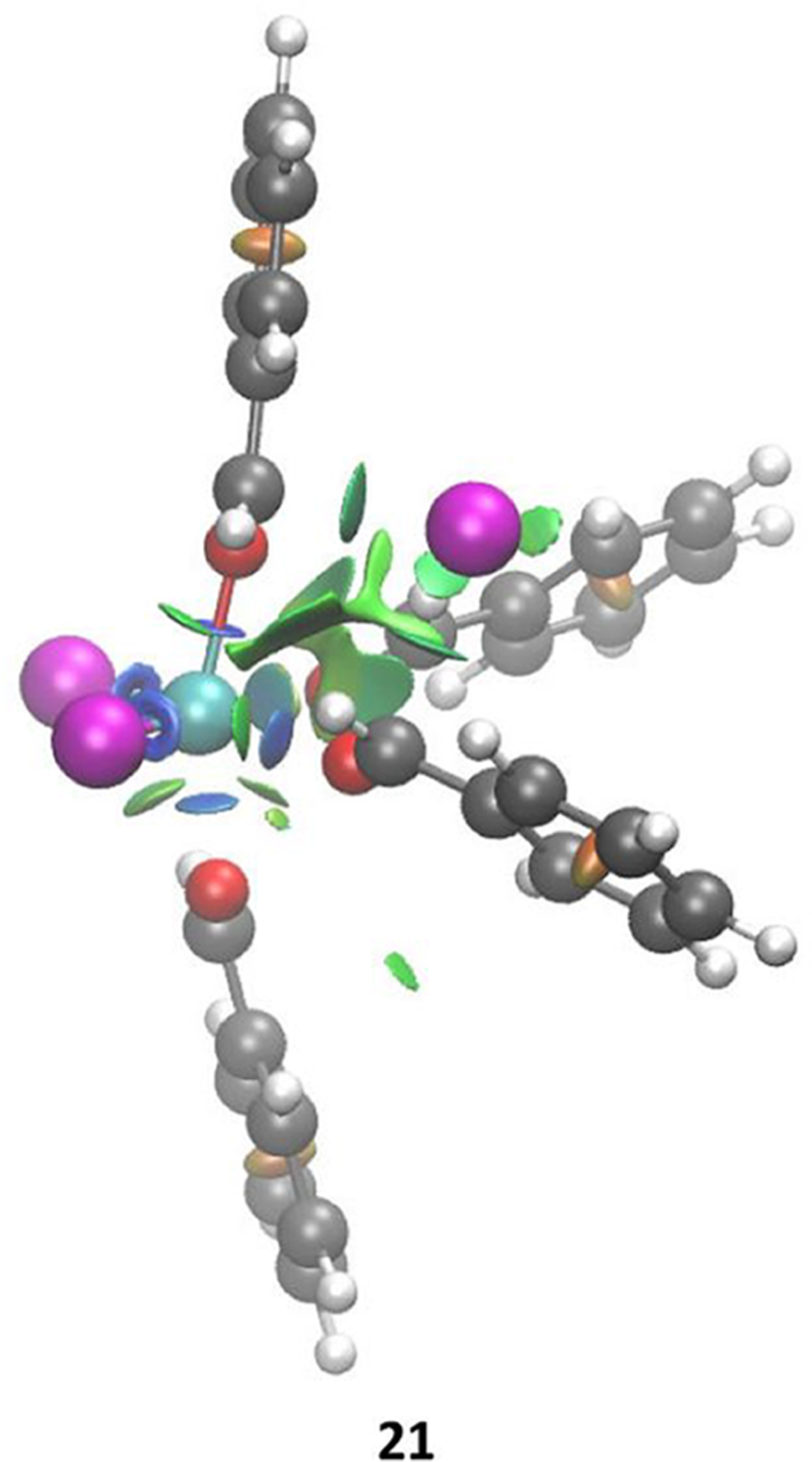
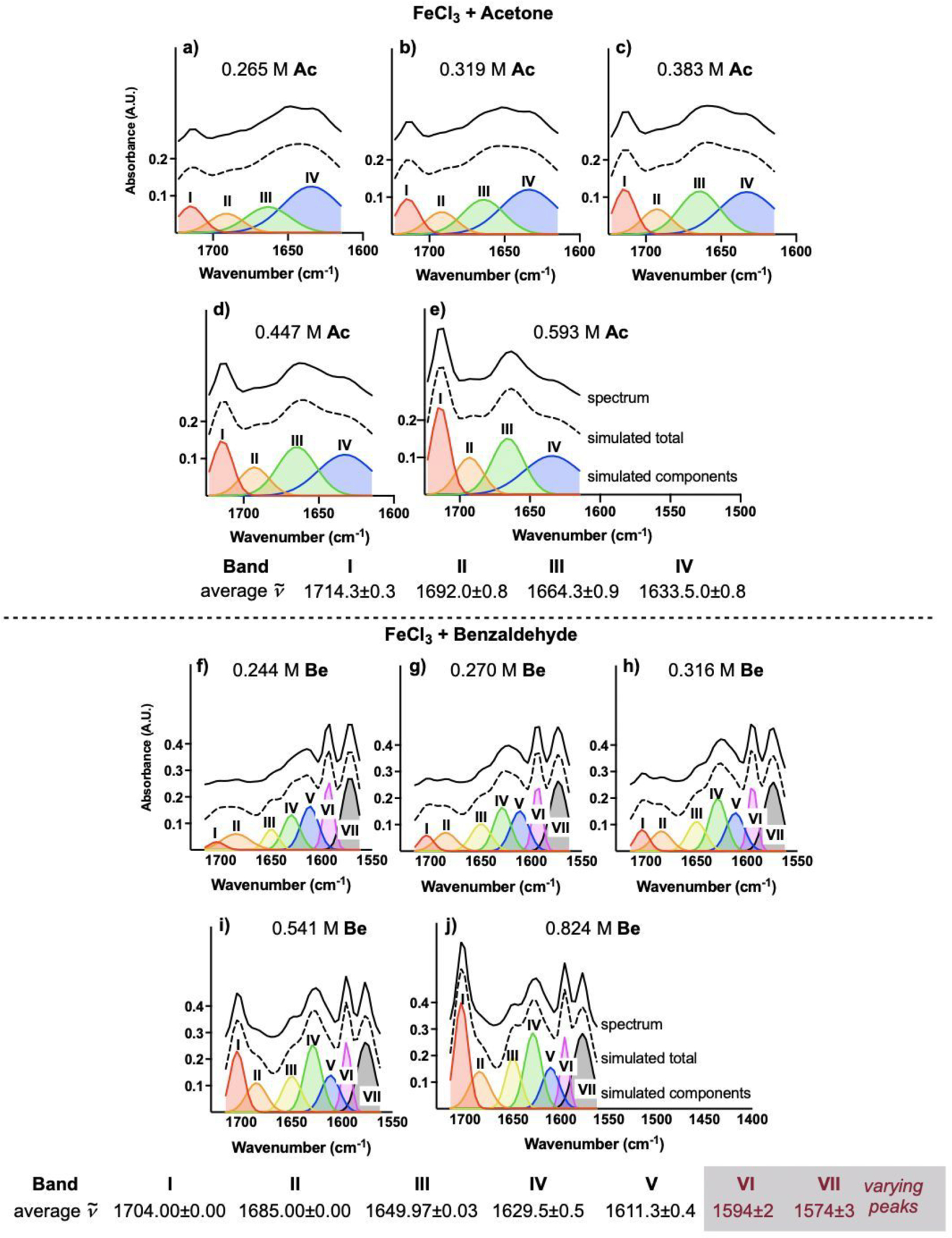
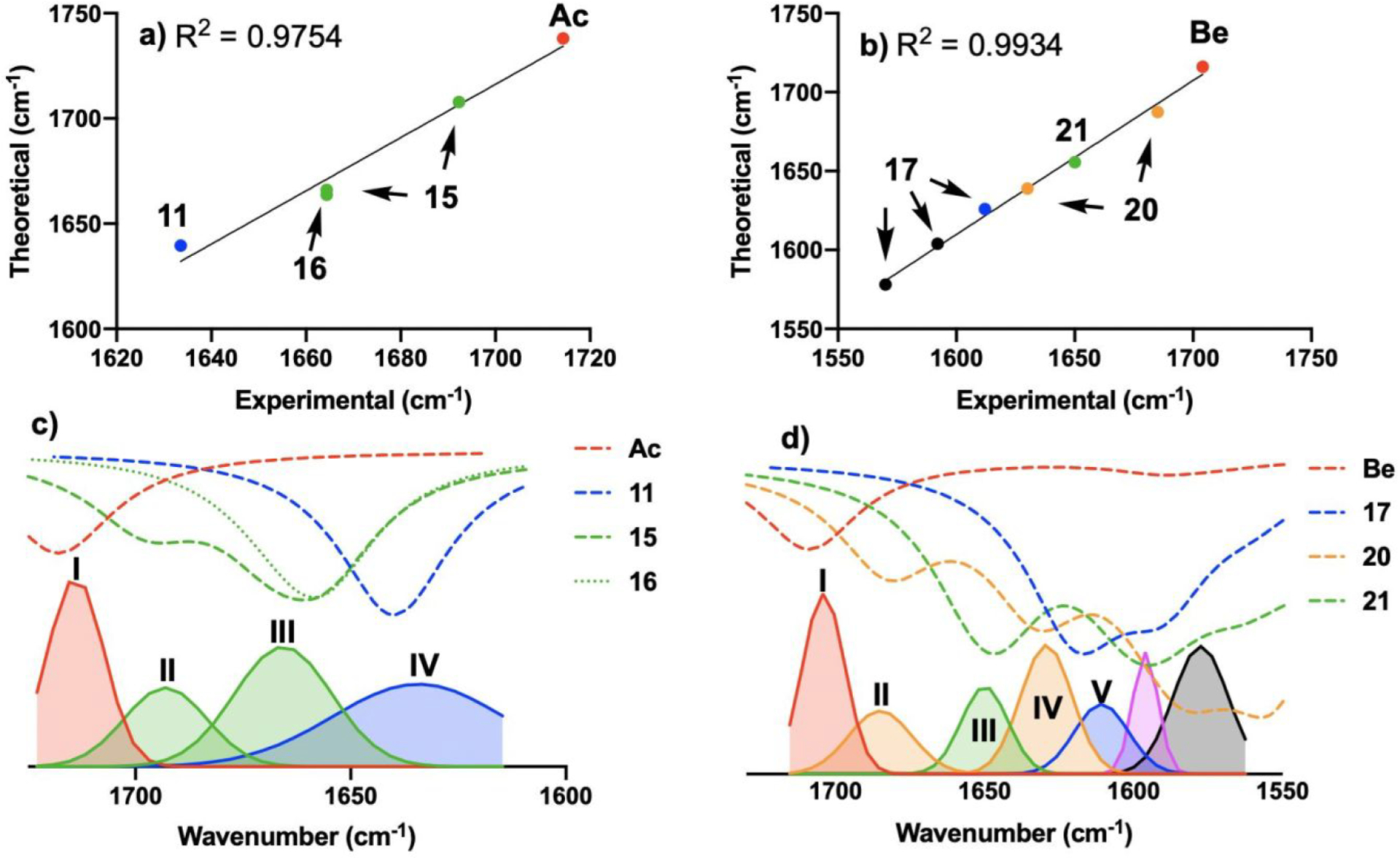
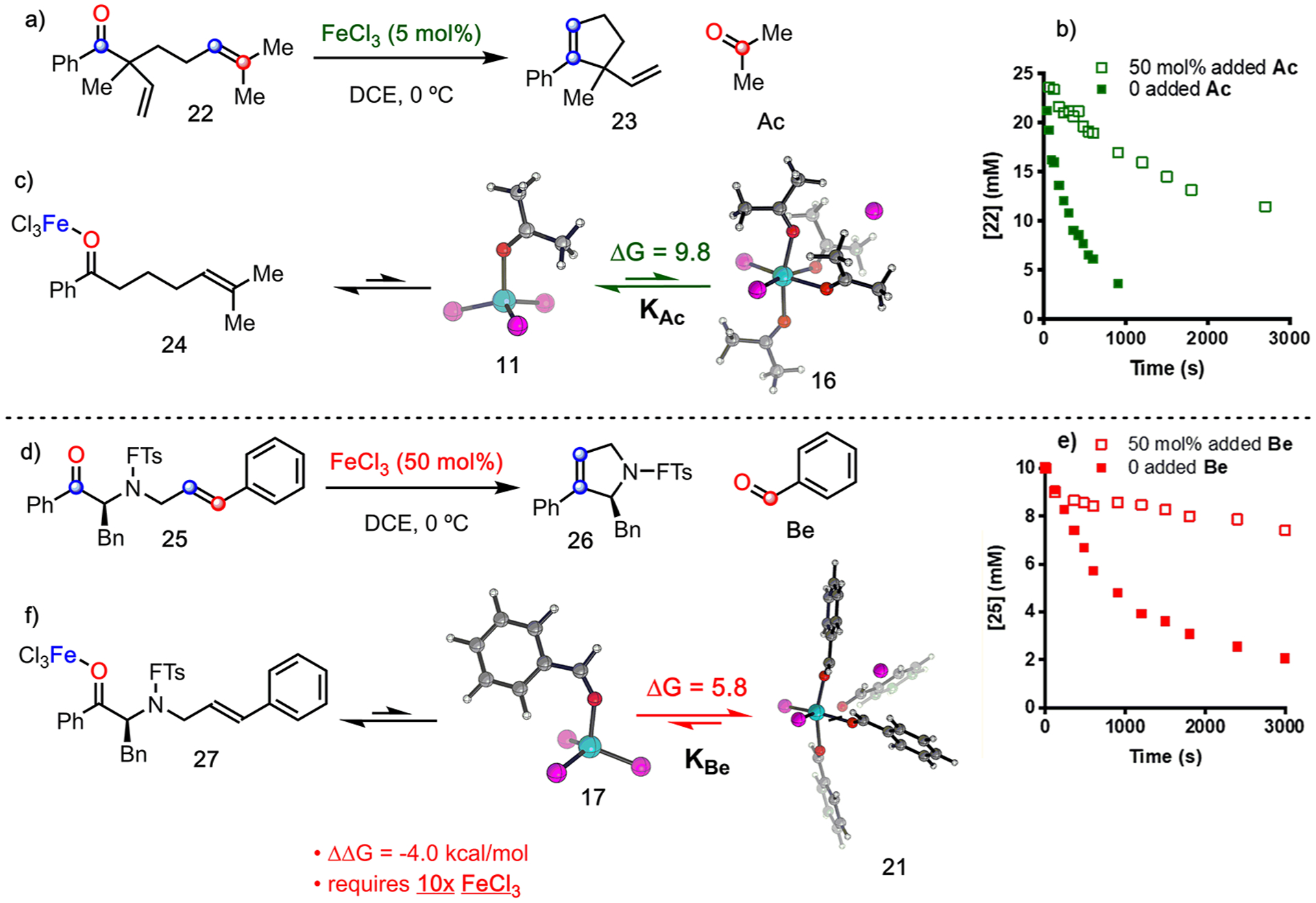
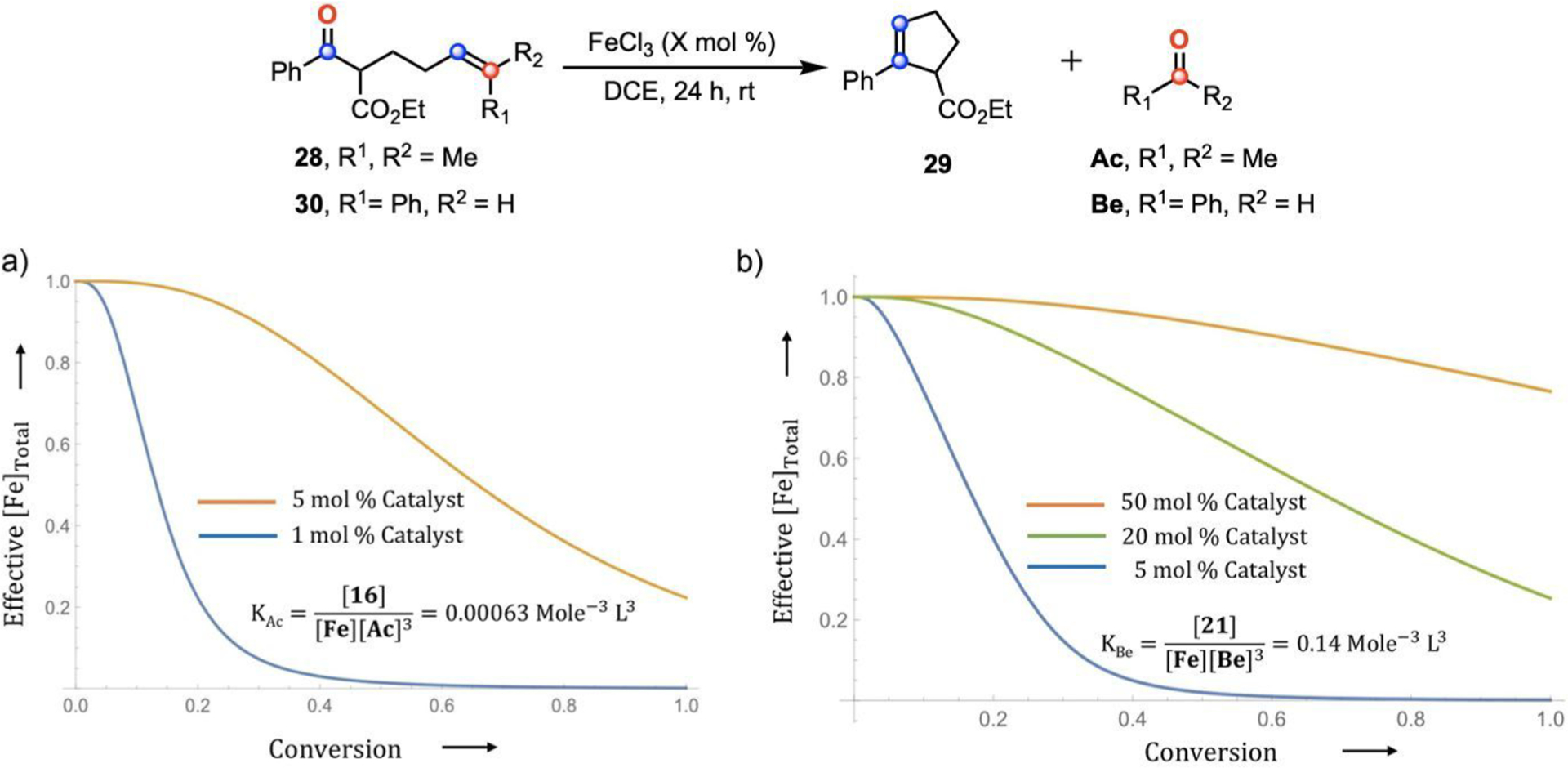
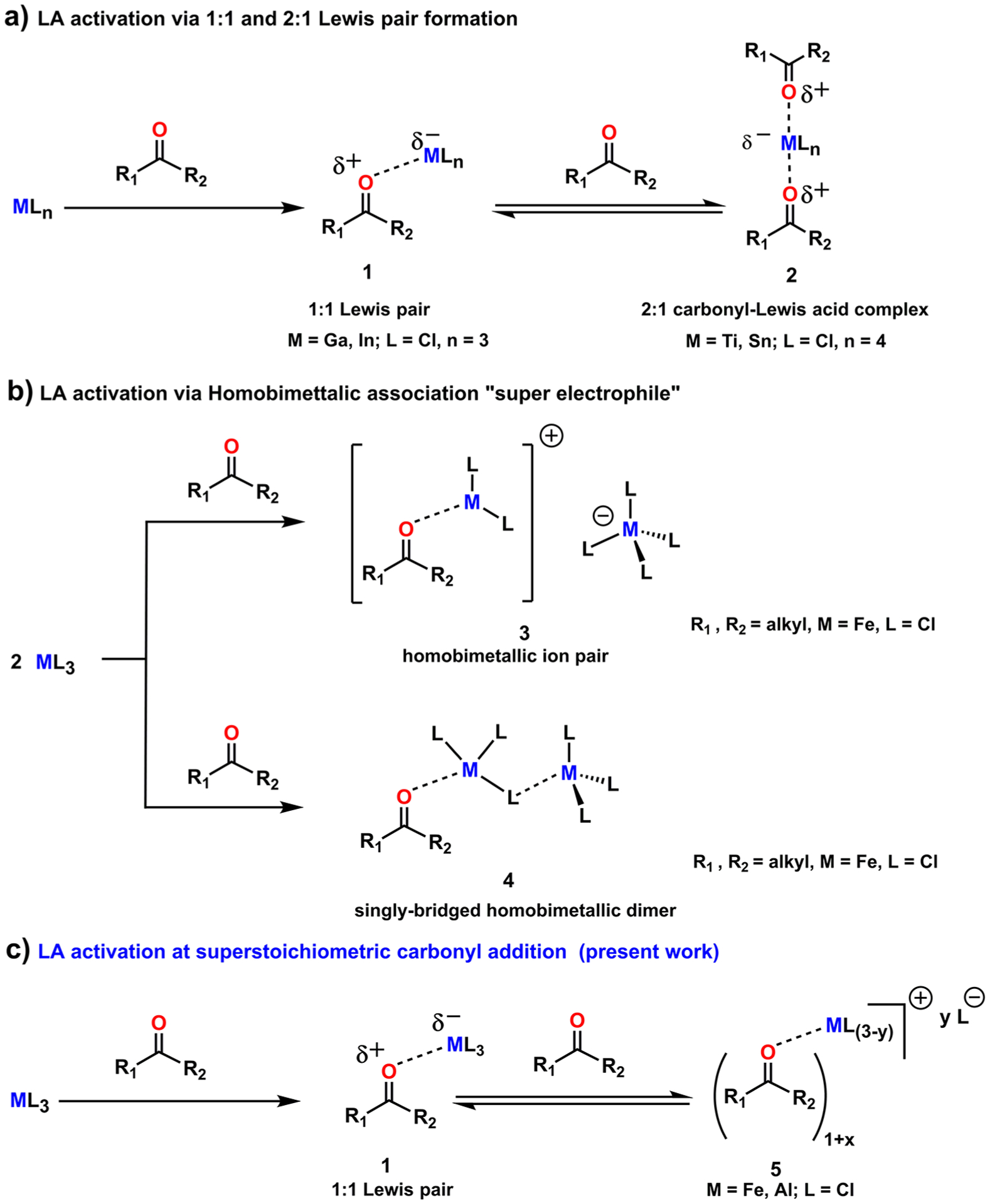
The coordination of a carbonyl to a Lewis acid represents the first step in a wide range of catalytic transformations. In many reactions it is necessary for the Lewis acid to discriminate between starting material and product, and as a result, how these structures behave in solution must be characterized. Herein, we report the application of computational modeling to calculate properties of the solution interactions of acetone and benzaldehyde with FeCl. Using these chemical models, we can predict spectral features in the carbonyl region of infrared (IR) spectroscopy. These simulated spectra are then directly compared to experimental spectra generated via titration-IR. We observe good agreement between theory and experiment, in that, between 0 and 1 equiv carbonyl with respect to FeCl, a pairwise interaction dominates the spectra. When >1 equiv carbonyl is present, our theoretical model predicts two possible structures composed of 4:1 carbonyl to FeCl, for acetone as well as benzaldehyde. When these predicted spectra are compared with titration-IR data, both structures contribute to the observed solution interactions. These findings suggest that the resting state of FeCl-catalyzed carbonyl-based reactions employing simple substrates starts as a Lewis pair, but this structure is gradually consumed and becomes a highly ligated, catalytically less active Fe-centered complex as the reaction proceeds. An analytical model is proposed to quantify catalyst inhibition due to equilibrium between 1:1 and 4:1 carbonyl:Fe complexes.
羰基与路易斯酸的配位是众多催化转化反应的第一步。在许多反应中,路易斯酸需要区分起始原料和产物,因此,必须表征这些结构在溶液中的行为。在此,我们报告了应用计算模型来计算丙酮和苯甲醛与FeCl在溶液中的相互作用性质。使用这些化学模型,我们可以预测红外(IR)光谱羰基区域的光谱特征。然后将这些模拟光谱直接与通过滴定红外产生的实验光谱进行比较。我们观察到理论与实验之间有很好的一致性,即在相对于FeCl为0至1当量羰基之间,成对相互作用主导光谱。当存在大于1当量羰基时,我们的理论模型预测了由丙酮和苯甲醛的羰基与FeCl比例为4:1组成的两种可能结构。当将这些预测光谱与滴定红外数据进行比较时,两种结构都对观察到的溶液相互作用有贡献。这些发现表明,使用简单底物的FeCl催化的基于羰基的反应的静止状态始于路易斯对,但随着反应进行,这种结构逐渐消耗并变成高度配位、催化活性较低的以铁为中心的配合物。提出了一个分析模型来量化由于1:1和4:1羰基:铁配合物之间的平衡导致的催化剂抑制作用。