Department of Neurology, Yongchuan Hospital of Chongqing Medical University, Chongqing 402160, China.
NHC Key Laboratory of Diagnosis and Treatment on Brain Functional Diseases, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, China.
J Adv Res. 2020 Dec 7;30:27-38. doi: 10.1016/j.jare.2020.12.002. eCollection 2021 May.
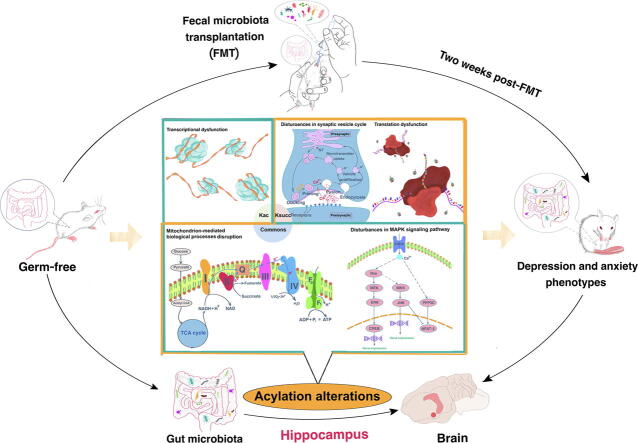
Major depressive disorder is caused by gene-environment interactions, and the host microbiome has been recognized as an important environmental factor. However, the underlying mechanisms of the host-microbiota interactions that lead to depression are complex and remain poorly understood.
The present study aimed to explore the possible mechanisms underlying gut microbiota dysbiosis-induced depressive-like behaviors.
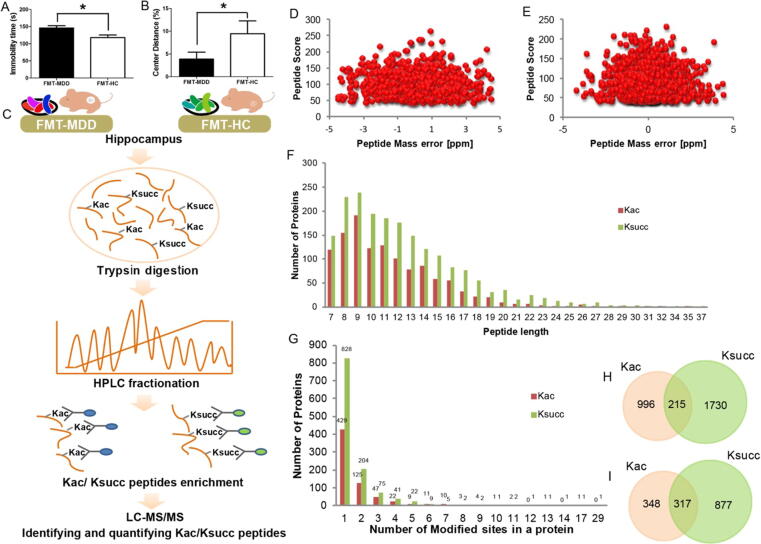
We used high-performance liquid chromatography-tandem mass spectrometry to analyze alterations in the hippocampal lysine acetylome and succinylome in male mice that had received gut microbiota from fecal samples of either patients with major depressive disorder or healthy controls. This was followed by bioinformatic analyses.
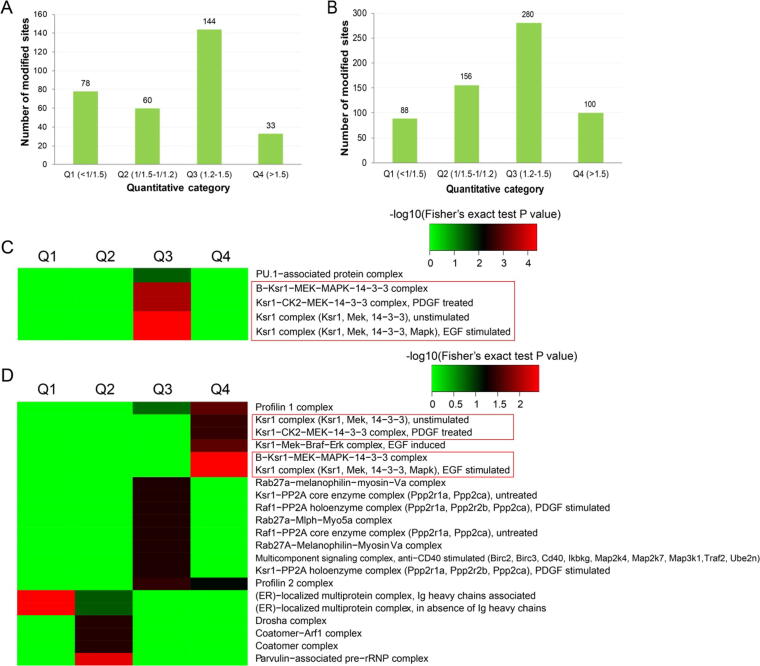
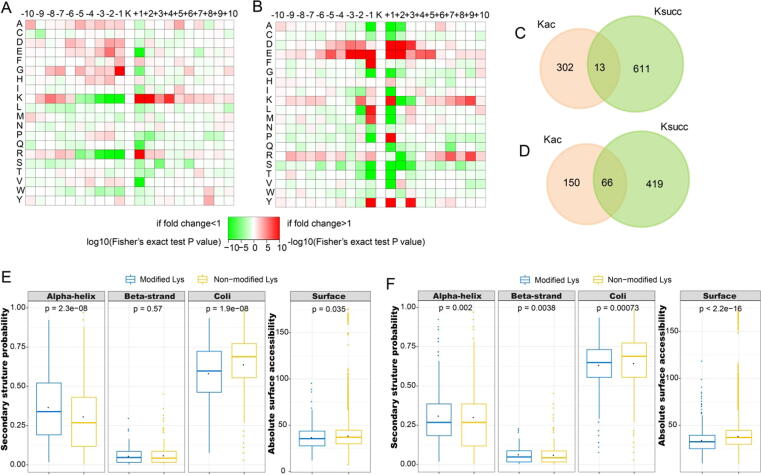
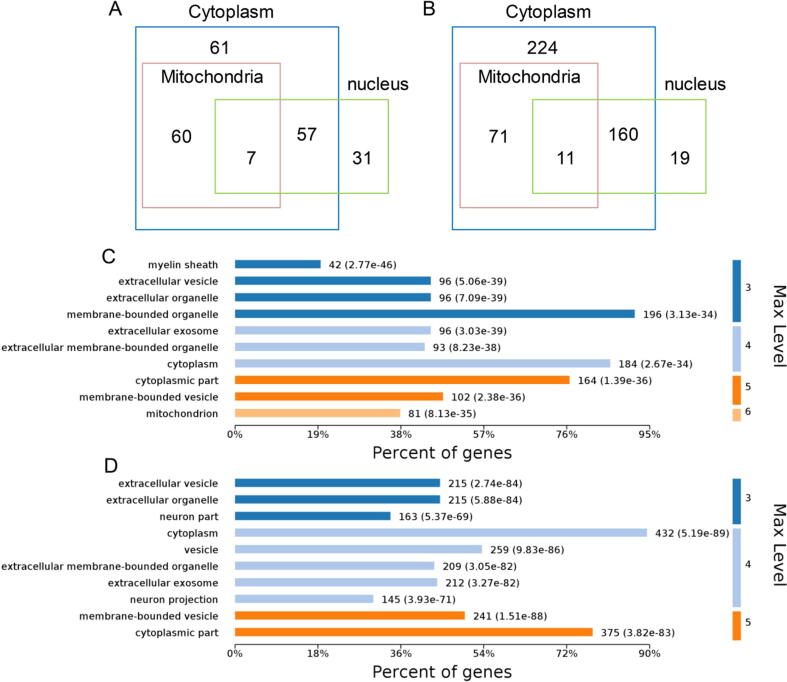
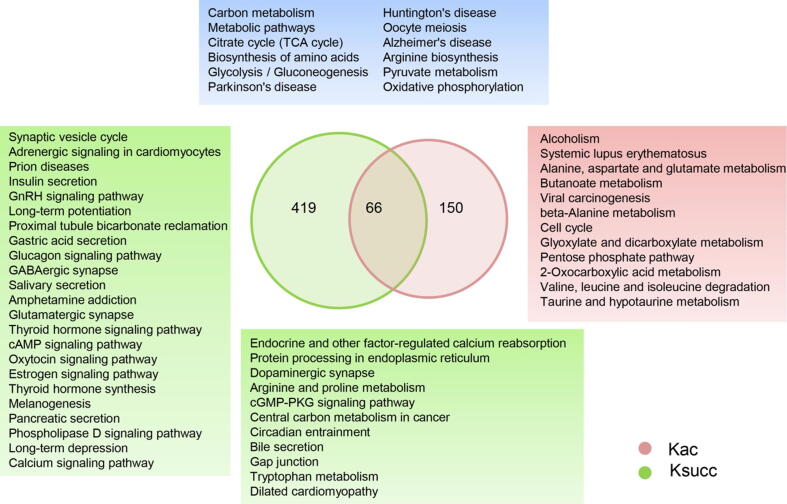
A total of 315 acetylation sites on 223 proteins and 624 succinylation sites on 494 proteins were differentially expressed in the gut microbiota-dysbiosis mice. The significantly acetylated proteins were primarily associated with carbon metabolism disruption and gene transcription suppression, while the synaptic vesicle cycle and protein translation were the most significantly altered functions for succinylated proteins. Additionally, our findings suggest that gut microbiota dysbiosis disturbs mitochondria-mediated biological processes and the MAPK signaling pathway through crosstalk between acetylation and succinylation on relevant proteins.
This is the first study to demonstrate modifications in acetylation and succinylation in gut microbiota-dysbiosis mice. Our findings provide new avenues for exploring the pathogenesis of gut microbiota dysbiosis-related depression, and highlight potential targets for depression treatment.
重度抑郁症是由基因-环境相互作用引起的,宿主微生物组已被认为是一个重要的环境因素。然而,导致抑郁的宿主-微生物群相互作用的潜在机制复杂且仍知之甚少。
本研究旨在探讨肠道微生物失调引起抑郁样行为的潜在机制。
我们使用高效液相色谱-串联质谱法分析来自重度抑郁症患者或健康对照的粪便样本的肠道微生物群的雄性小鼠海马体赖氨酸乙酰组和琥珀酰组的变化。随后进行生物信息学分析。
在肠道微生物失调的小鼠中,共有 223 种蛋白质的 315 个乙酰化位点和 494 种蛋白质的 624 个琥珀酰化位点发生了差异表达。显著乙酰化的蛋白质主要与碳代谢紊乱和基因转录抑制有关,而突触囊泡循环和蛋白质翻译是琥珀酰化蛋白质最显著改变的功能。此外,我们的研究结果表明,肠道微生物失调通过相关蛋白质的乙酰化和琥珀酰化之间的串扰扰乱了线粒体介导的生物学过程和 MAPK 信号通路。
这是第一项研究表明在肠道微生物失调的小鼠中存在乙酰化和琥珀酰化修饰的研究。我们的研究结果为探索与肠道微生物失调相关的抑郁症的发病机制提供了新的途径,并强调了治疗抑郁症的潜在靶点。