Capital Institute of Pediatrics, 2 yabao road, Chaoyang District, Beijing, China.
Pediatr Rheumatol Online J. 2021 May 24;19(1):75. doi: 10.1186/s12969-021-00558-6.
To evaluate the clinical and genetic characteristics of 3 children with Haploinsufficiency of A20 (HA20).
The clinical and genetic testing data of 3 children with HA20 treated at Capital Institute of Pediatrics (CIP) between August 2016 and October 2019 were retrospectively analysed.
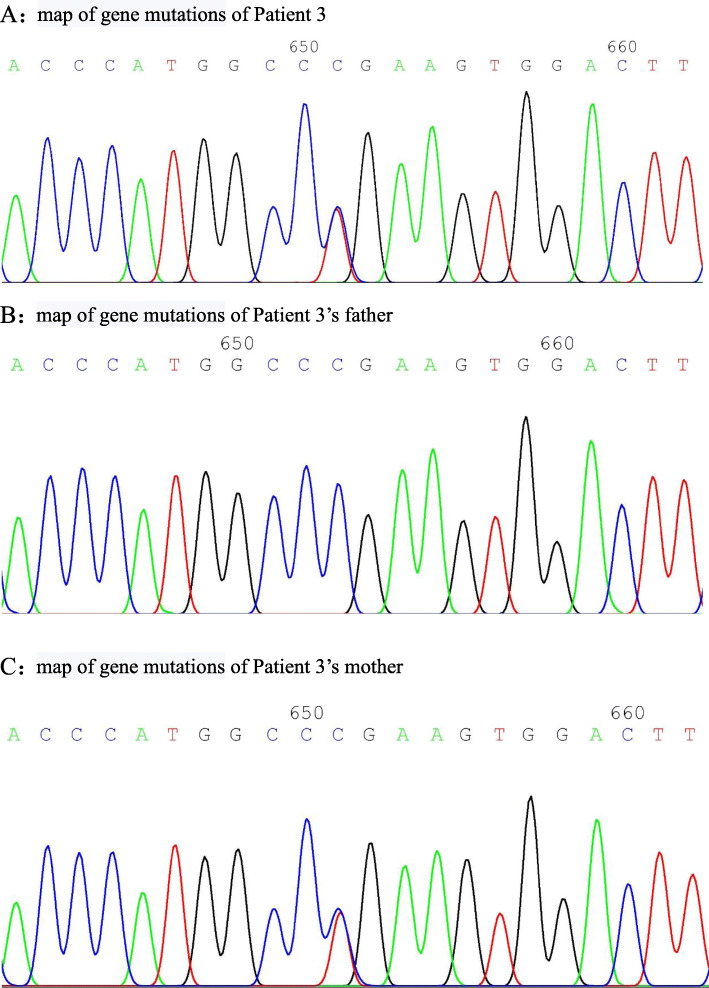
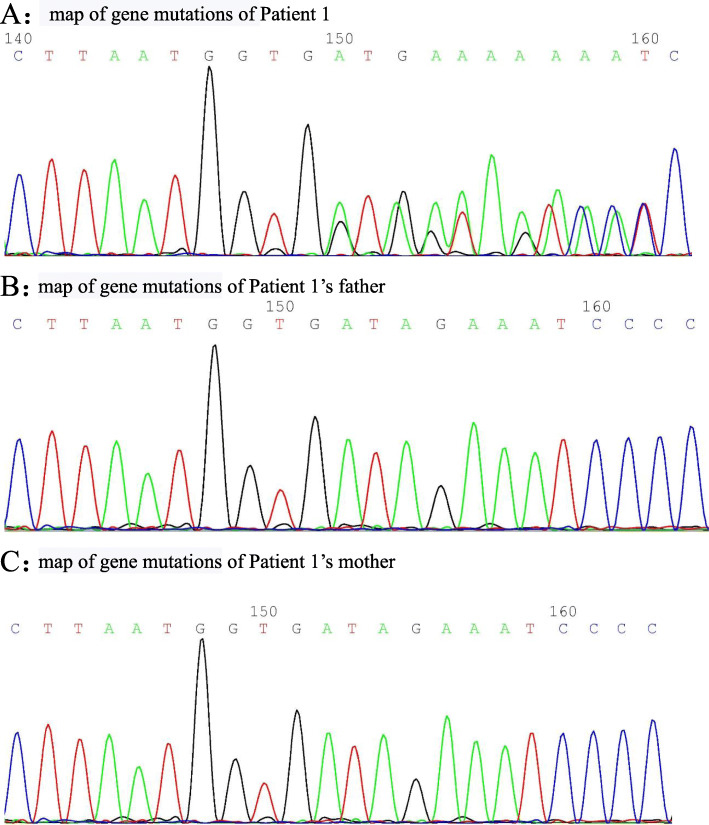
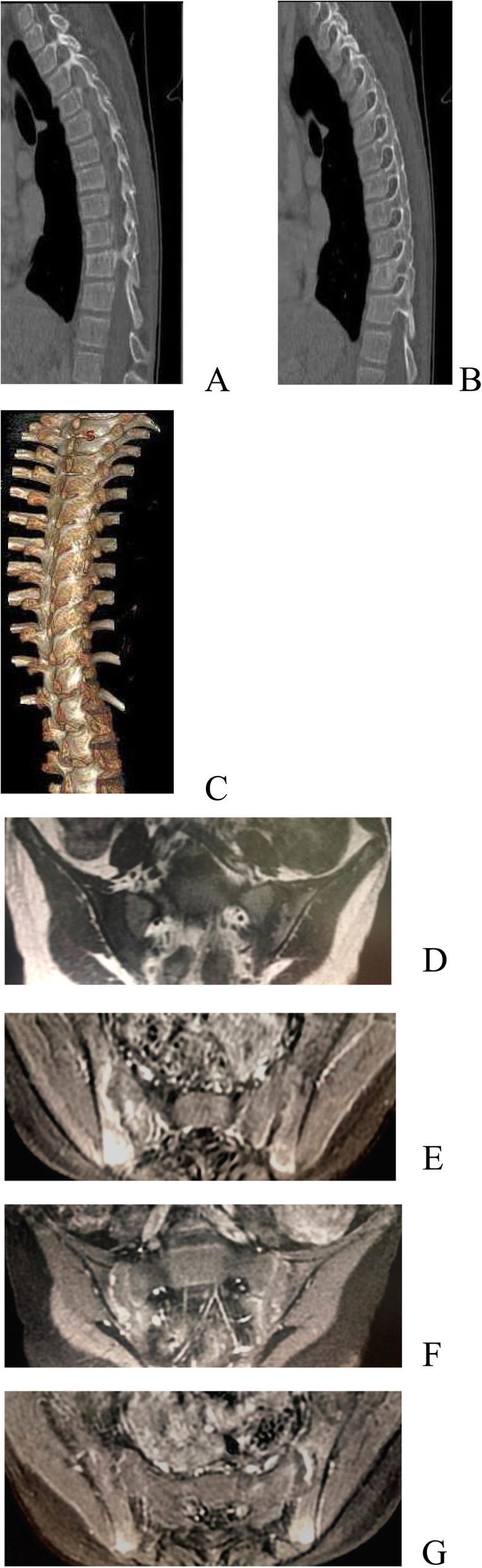
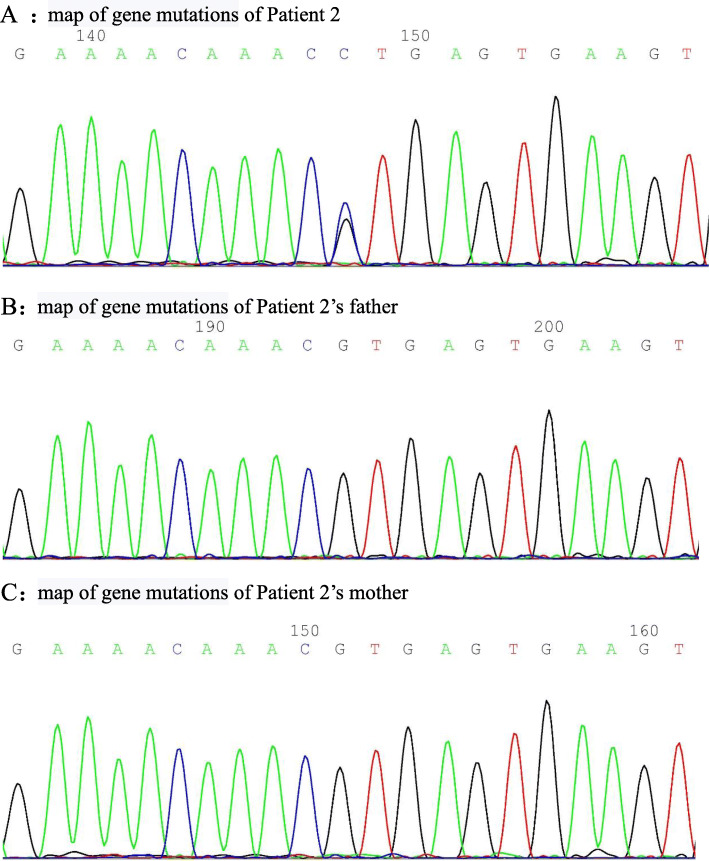
Patient 1 presented with arthritis and inflammatory bowel disease, patient 2 presented with axial spinal arthritis and lupus-like syndrome, and patient 3 presented with recurrent oral ulcers, gastrointestinal ulcers, and perianal abscesses. Regarding laboratory tests, patients were found to have elevated white blood cell (WBC) count, C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR). The CRP and ESR was reported to be high in all the patients. The WBC was reported to be high in patient 1 and 3. Patient 2 was positive for antinuclear antibodies, anti-Sjögren's syndrome antigen A, dsDNA, rheumatoid factor and Coombs test. Genetic testing showed that all three patients had heterozygous mutation in TNFAIP3 gene. As for the treatment, patient 1 was treated with TNFα antagonist, patient 2 was treated with TNF α antagonist and sulfasalazine, and patient 3 was treated with corticosteroids and thalidomide. Patients 1 and 2 were followed for four and 3 months, respectively. There was an improvement in joint and gastrointestinal symptoms; inflammatory indices and rheumatoid factor (RF) were normal, and dsDNA and Coombs test became negative. Patient 3 was treated at another hospital and showed gradual improvement in oral ulcers and perianal abscesses.
HA20 is a single-gene auto-inflammatory disease caused by mutation in tumour necrosis factor (TNF)-α-induced protein 3 (TNFAIP3) gene. It may present as Behçet-like syndrome and resemble various other autoimmune diseases as well. Corticosteroids and immunosuppressive agents are effective treatments, and cytokine antagonists can be used in refractory cases. Whole-exome genetic testing should be proactively performed for children with early-age onset or Behçet-like syndrome to achieve early diagnosis and accurate treatment.
评估 3 例 A20 单倍体不足(HA20)患儿的临床和遗传特征。
回顾性分析首都儿科研究所 2016 年 8 月至 2019 年 10 月收治的 3 例 HA20 患儿的临床和基因检测资料。
患儿 1 表现为关节炎和炎症性肠病,患儿 2 表现为轴性脊柱关节炎和狼疮样综合征,患儿 3 表现为复发性口腔溃疡、胃肠道溃疡和肛周脓肿。实验室检查发现,患儿白细胞(WBC)计数、C 反应蛋白(CRP)和红细胞沉降率(ESR)升高,所有患儿 CRP 和 ESR 均高,WBC 计数仅患儿 1 和 3 升高。患儿 2 抗核抗体、抗干燥综合征 A 抗原、双链 DNA、类风湿因子和抗球蛋白试验阳性。基因检测发现 3 例患儿均存在 TNFAIP3 基因杂合突变。治疗方面,患儿 1 给予 TNFα 拮抗剂治疗,患儿 2 给予 TNFα 拮抗剂和柳氮磺胺吡啶治疗,患儿 3 给予糖皮质激素和沙利度胺治疗。患儿 1 和 2 分别随访 4 个月和 3 个月,关节和胃肠道症状改善,炎症指标和类风湿因子(RF)正常,双链 DNA 和抗球蛋白试验转阴。患儿 3 在其他医院治疗,口腔溃疡和肛周脓肿逐渐好转。
HA20 是一种由肿瘤坏死因子(TNF)-α诱导蛋白 3(TNFAIP3)基因突变引起的单基因自身炎症性疾病,可表现为贝赫切特样综合征,也可类似其他多种自身免疫性疾病。糖皮质激素和免疫抑制剂治疗有效,对难治性病例可使用细胞因子拮抗剂。对于早发型或贝赫切特样综合征患儿,应积极行全外显子组基因检测,以实现早期诊断和精准治疗。