Conde Cyril, Price-Carter Marian, Cochard Thierry, Branger Maxime, Stevenson Karen, Whittington Richard, Bannantine John P, Biet Franck
INRAE, ISP, Université de Tours, Nouzilly, France.
AgResearch Ltd., Hopkirk Research Institute, Palmerston North, New Zealand.
Front Microbiol. 2021 May 10;12:660002. doi: 10.3389/fmicb.2021.660002. eCollection 2021.
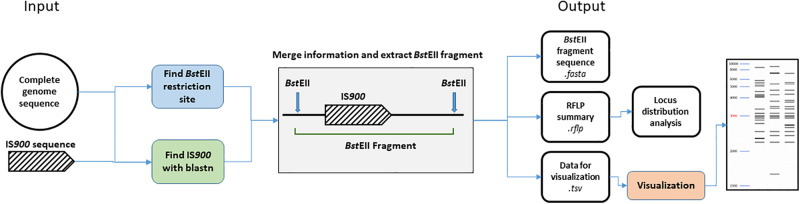
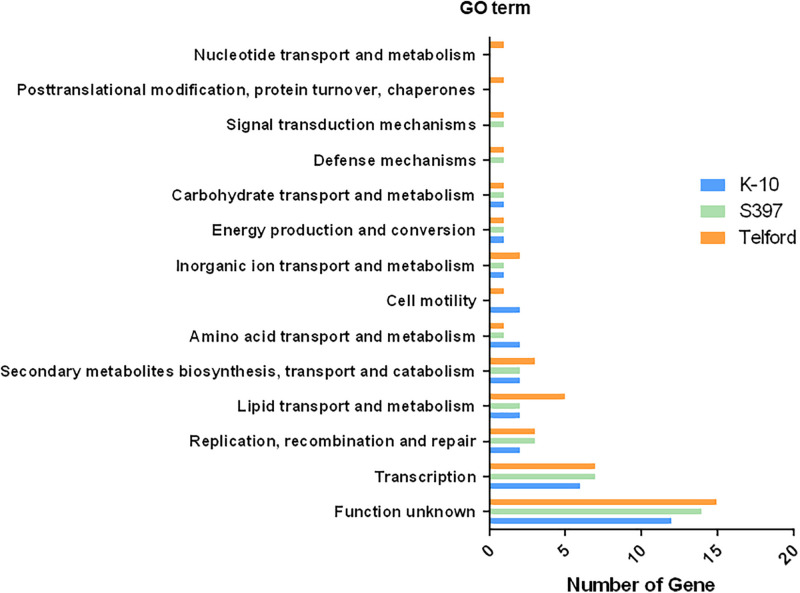
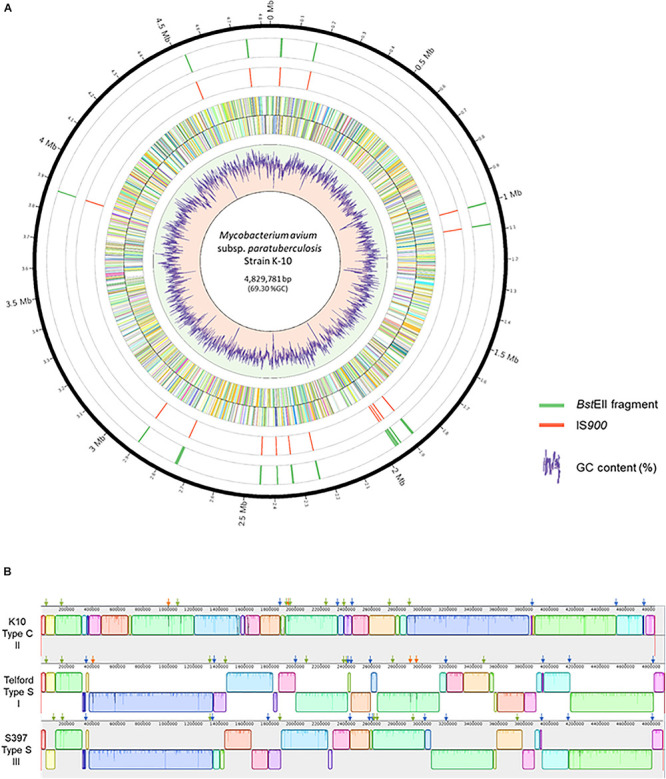
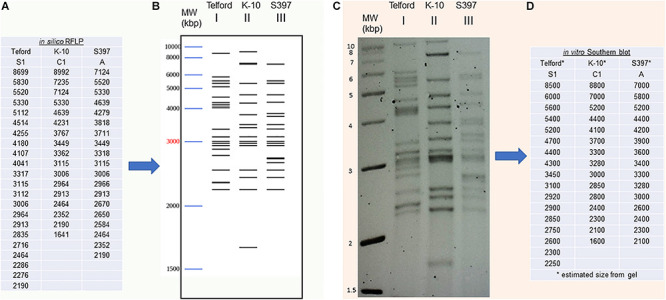
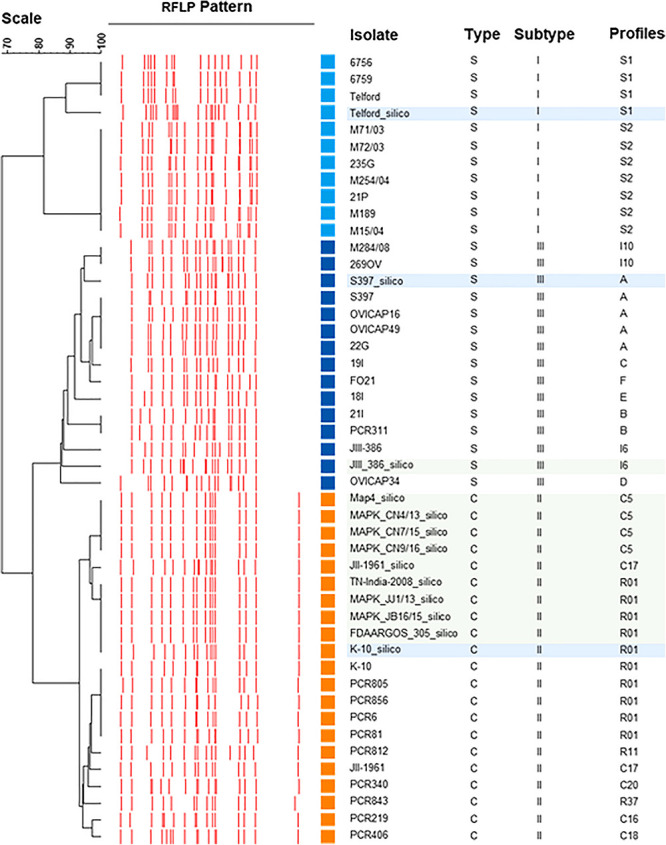
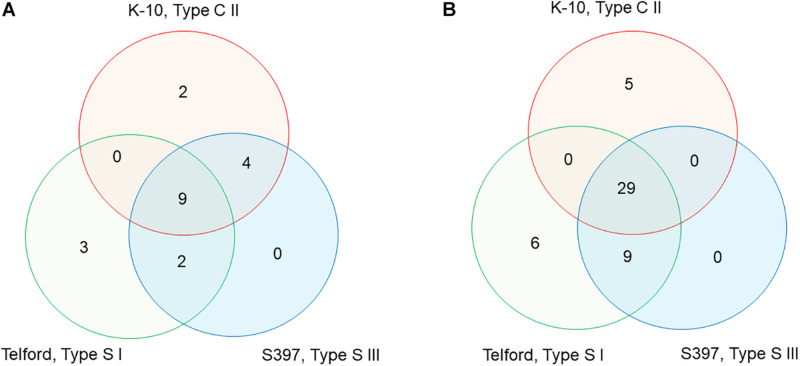
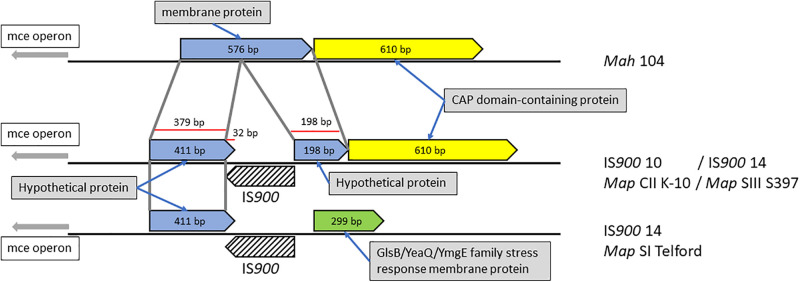
subsp. () is the etiological agent of Johne's disease in ruminants. The IS insertion sequence (IS) has been used widely as an epidemiological marker and target for PCR diagnosis. Updated DNA sequencing technologies have led to a rapid increase in available genomes, which makes it possible to analyze the distribution of IS in this slow-growing bacterium. The objective of this study is to characterize the distribution of the IS element and how it affects genomic evolution and gene function of . A secondary goal is to develop automated restriction fragment length polymorphism (RFLP) analysis using IS. Complete genomes from the major phylogenetic lineages known as C-type and S-type (including subtypes I and III), were chosen to represent the genetic diversity of . IS elements were located in these genomes using BLAST software and the relevant fragments extracted. An RFLP analysis using the EII restriction site was performed to obtain exact sizes of the DNA fragments carrying a copy of IS and the resulting RFLP profiles were analyzed and compared by digital visualization of the separated restriction fragments. The program developed for this study allowed automated localization of IS sequences to identify their position within each genome along with the exact number of copies per genome. The number of IS copies ranged from 16 in the C-type isolate to 22 in the S-type subtype I isolate. A loci-by-loci sequence alignment of all IS copies within the three genomes revealed new sequence polymorphisms that define three sequevars distinguishing the subtypes. Nine IS insertion site locations were conserved across all genomes studied while smaller subsets were unique to a particular lineage. Preferential insertion motif sequences were identified for IS along with genes bordering all IS insertions. Rarely did IS insert within coding sequences as only three genes were disrupted in this way. This study makes it possible to automate IS distribution in genomes to enrich knowledge on the distribution dynamics of this IS for epidemiological purposes, for understanding evolution and for studying the biological implications of IS insertions.
亚种( )是反刍动物约翰氏病的病原体。插入序列(IS)已被广泛用作流行病学标记和PCR诊断的靶标。更新的DNA测序技术使可用基因组数量迅速增加,这使得分析这种生长缓慢的细菌中IS的分布成为可能。本研究的目的是表征IS元件的分布及其如何影响 的基因组进化和基因功能。第二个目标是开发使用IS的自动化 限制性片段长度多态性(RFLP)分析。选择来自已知的C型和S型(包括亚型I和III)主要系统发育谱系的完整基因组来代表 的遗传多样性。使用BLAST软件在这些基因组中定位IS元件并提取相关片段。使用EII限制性位点进行 RFLP分析,以获得携带IS拷贝的DNA片段的精确大小,并通过对分离的限制性片段进行数字可视化来分析和比较所得的RFLP图谱。为本研究开发的程序允许自动定位IS序列,以确定它们在每个基因组中的位置以及每个基因组的确切拷贝数。IS拷贝数范围从C型分离株中的16个到S型亚型I分离株中的22个。对三个基因组中所有IS拷贝进行逐个位点的序列比对,揭示了新的序列多态性,这些多态性定义了区分亚型的三个序列变种。在所有研究的基因组中,九个IS插入位点位置是保守的,而较小的子集是特定谱系所特有的。确定了IS的优先插入基序序列以及所有IS插入旁边的基因。IS很少插入编码序列中,因为只有三个基因以这种方式被破坏。本研究使得自动分析 基因组中的IS分布成为可能,以丰富关于这种IS分布动态的知识,用于流行病学目的、理解 的进化以及研究IS插入的生物学意义。