Department of Biology, University of Washington, Seattle, WA, USA.
Computational Biology Program, Fred Hutchinson Cancer Research Center, Seattle, WA, USA.
Mol Biol Evol. 2021 Sep 27;38(10):4603-4615. doi: 10.1093/molbev/msab163.
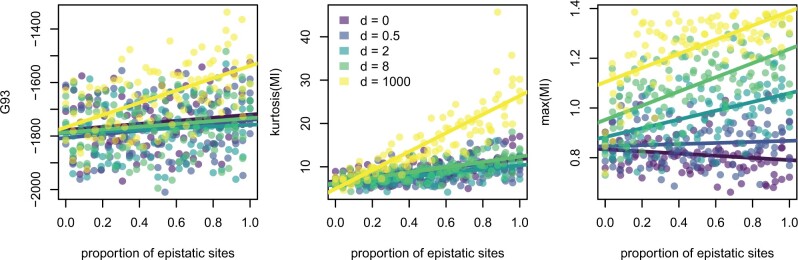
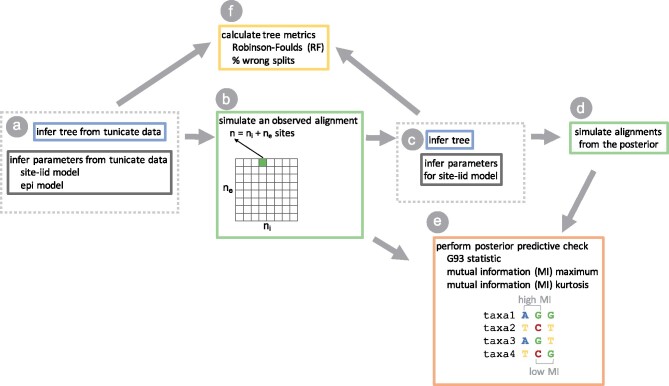
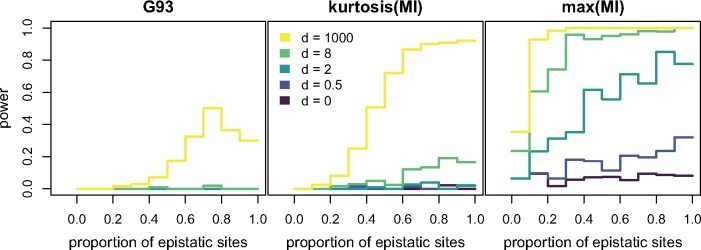
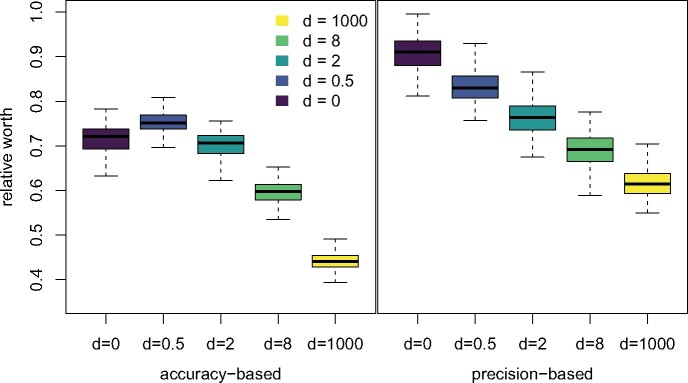
Likelihood-based phylogenetic inference posits a probabilistic model of character state change along branches of a phylogenetic tree. These models typically assume statistical independence of sites in the sequence alignment. This is a restrictive assumption that facilitates computational tractability, but ignores how epistasis, the effect of genetic background on mutational effects, influences the evolution of functional sequences. We consider the effect of using a misspecified site-independent model on the accuracy of Bayesian phylogenetic inference in the setting of pairwise-site epistasis. Previous work has shown that as alignment length increases, tree reconstruction accuracy also increases. Here, we present a simulation study demonstrating that accuracy increases with alignment size even if the additional sites are epistatically coupled. We introduce an alignment-based test statistic that is a diagnostic for pairwise epistasis and can be used in posterior predictive checks.
基于可能性的系统发育推断假设了一种沿系统发育树分支的字符状态变化的概率模型。这些模型通常假设序列比对中各位置的统计独立性。这是一个限制性假设,它便于计算,但忽略了遗传背景对突变效应的影响(上位性)如何影响功能序列的进化。我们考虑了在成对位置上位性的情况下,使用不合适的不依赖于位置的模型对贝叶斯系统发育推断准确性的影响。以前的工作表明,随着对齐长度的增加,树重建的准确性也会提高。在这里,我们进行了一项模拟研究,表明即使附加的位置是上位性耦合的,准确性也会随着对齐大小的增加而增加。我们引入了一个基于对齐的检验统计量,它是一种用于成对上位性的诊断方法,可以用于后验预测检查。