Letiagina Anna E, Omelina Evgeniya S, Ivankin Anton V, Pindyurin Alexey V
Institute of Molecular and Cellular Biology of the Siberian Branch of the Russian Academy of Sciences, Novosibirsk, Russia.
Faculty of Natural Sciences, Novosibirsk State University, Novosibirsk, Russia.
Front Genet. 2021 May 11;12:618189. doi: 10.3389/fgene.2021.618189. eCollection 2021.
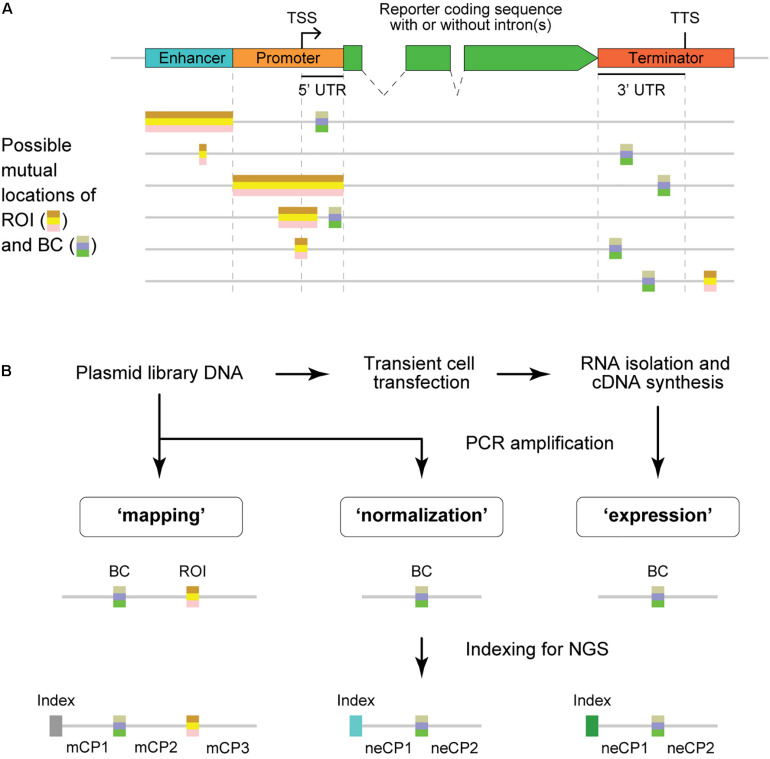
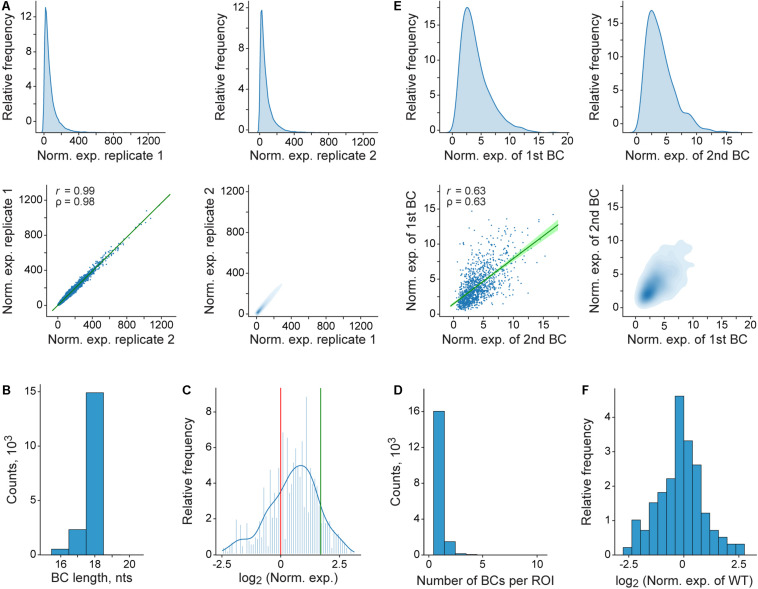
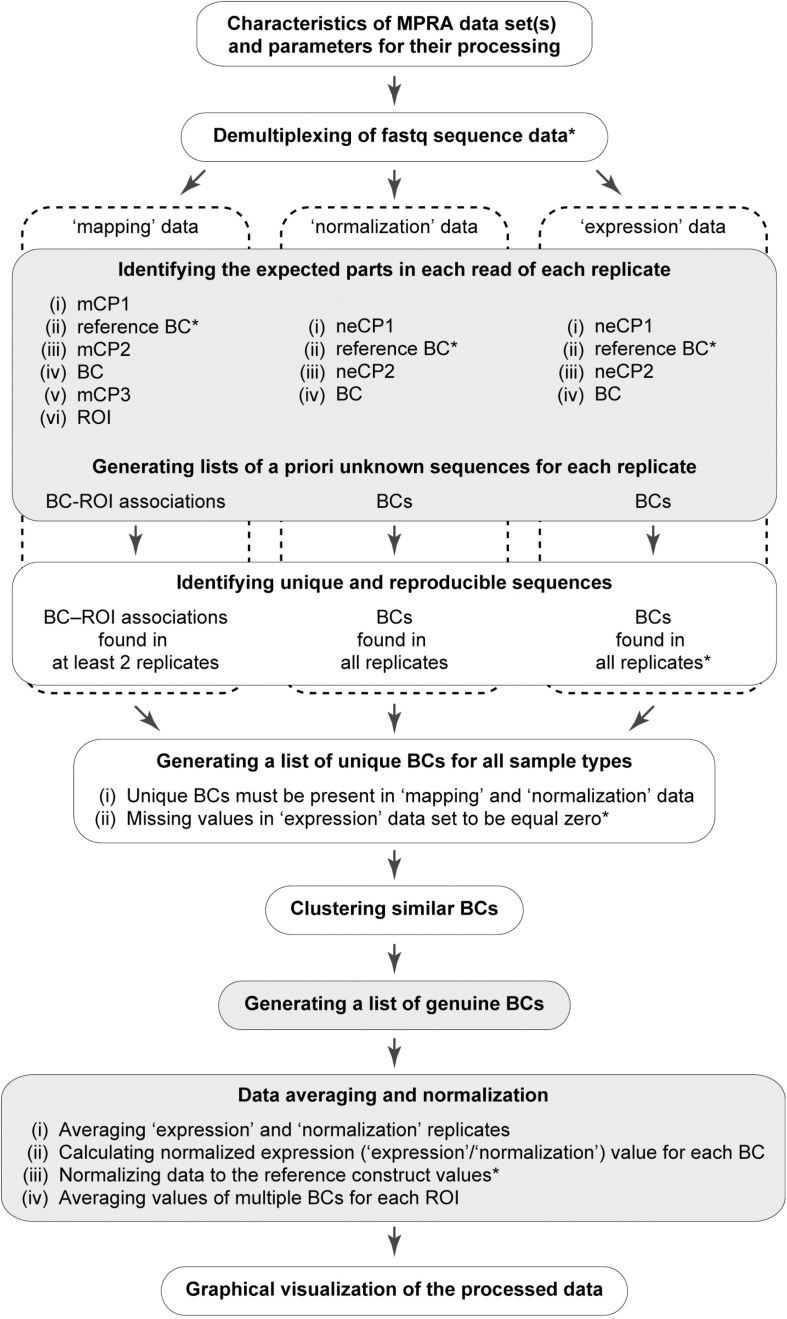
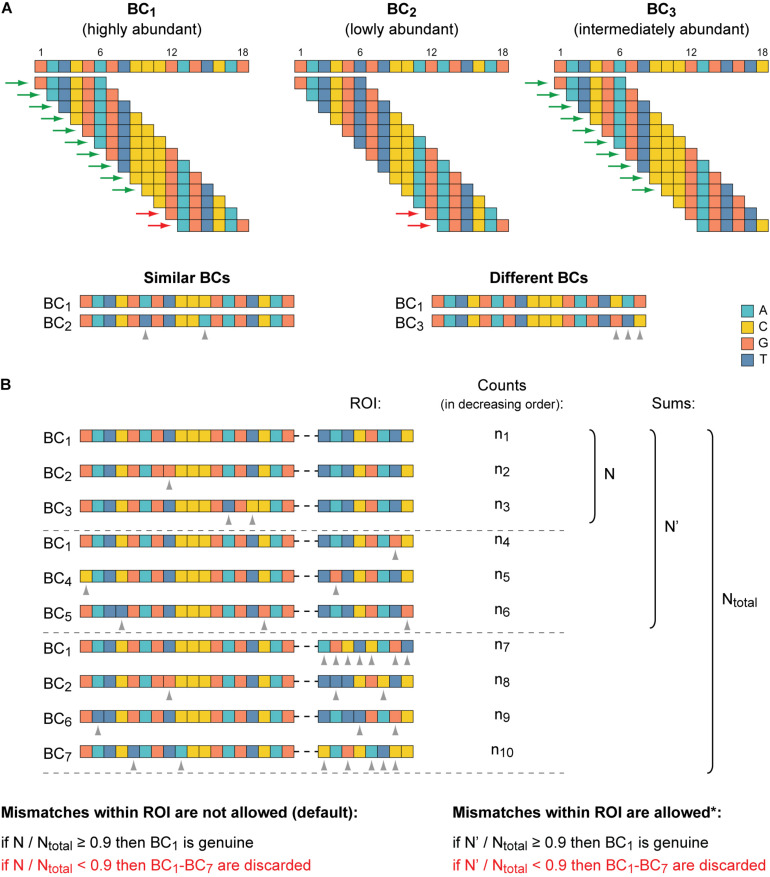
Massively parallel reporter assays (MPRAs) enable high-throughput functional evaluation of numerous DNA regulatory elements and/or their mutant variants. The assays are based on the construction of reporter plasmid libraries containing two variable parts, a region of interest (ROI) and a barcode (BC), located outside and within the transcription unit, respectively. Importantly, each plasmid molecule in a such a highly diverse library is characterized by a unique BC-ROI association. The reporter constructs are delivered to target cells and expression of BCs at the transcript level is assayed by RT-PCR followed by next-generation sequencing (NGS). The obtained values are normalized to the abundance of BCs in the plasmid DNA sample. Altogether, this allows evaluating the regulatory potential of the associated ROI sequences. However, depending on the MPRA library construction design, the BC and ROI sequences as well as their associations can be unknown. In such a case, the BC and ROI sequences, their possible mutant variants, and unambiguous BC-ROI associations have to be identified, whereas all uncertain cases have to be excluded from the analysis. Besides the preparation of additional "mapping" samples for NGS, this also requires specific bioinformatics tools. Here, we present a pipeline for processing raw MPRA data obtained by NGS for reporter construct libraries with unknown sequences of BCs and ROIs. The pipeline robustly identifies unambiguous (so-called genuine) BCs and ROIs associated with them, calculates the normalized expression level for each BC and the averaged values for each ROI, and provides a graphical visualization of the processed data.
大规模平行报告基因检测(MPRAs)能够对众多DNA调控元件及其突变变体进行高通量功能评估。这些检测基于构建报告质粒文库,该文库包含两个可变部分,分别是位于转录单元外部的感兴趣区域(ROI)和位于转录单元内部的条形码(BC)。重要的是,如此高度多样化的文库中的每个质粒分子都具有独特的BC-ROI关联特征。将报告构建体导入靶细胞,通过逆转录聚合酶链反应(RT-PCR)随后进行下一代测序(NGS)来检测转录水平上BC的表达。将获得的值归一化为质粒DNA样品中BC的丰度。总之,这使得能够评估相关ROI序列调控潜力。然而,根据MPRA文库构建设计,BC和ROI序列及其关联可能是未知的。在这种情况下,必须识别BC和ROI序列、它们可能的突变变体以及明确的BC-ROI关联,而所有不确定的情况都必须从分析中排除。除了为NGS制备额外的“定位”样品外,这还需要特定的生物信息学工具。在这里,我们提出了一个流程,用于处理通过NGS获得的、针对BC和ROI序列未知的报告构建体文库的原始MPRA数据。该流程能够可靠地识别与它们相关的明确(即所谓的真实)BC和ROI,计算每个BC的归一化表达水平以及每个ROI的平均值,并提供处理后数据的图形可视化。