Institute of Molecular and Cellular Biology SB RAS, Novosibirsk, Russia.
Novosibirsk State University, Novosibirsk, Russia.
BMC Genomics. 2019 Jul 11;20(Suppl 7):536. doi: 10.1186/s12864-019-5847-2.
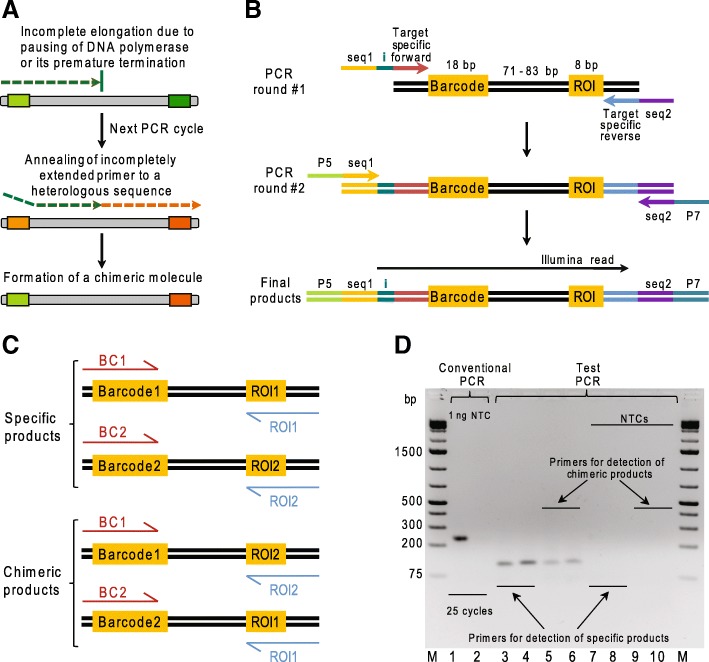
Massively parallel reporter assays (MPRAs) enable high-throughput functional evaluation of various DNA regulatory elements and their mutant variants. The assays are based on construction of highly diverse plasmid libraries containing two variable fragments, a region of interest (a sequence under study; ROI) and a barcode (BC) used to uniquely tag each ROI, which are separated by a constant spacer sequence. The sequences of BC-ROI combinations present in the libraries may be either known a priori or not. In the latter case, it is necessary to identify these combinations before performing functional experiments. Typically, this is done by PCR amplification of the BC-ROI regions with flanking primers, followed by next-generation sequencing (NGS) of the products. However, chimeric DNA molecules formed on templates with identical spacer fragment during the amplification process may substantially hamper the identification of genuine BC-ROI combinations, and as a result lower the performance of the assays.
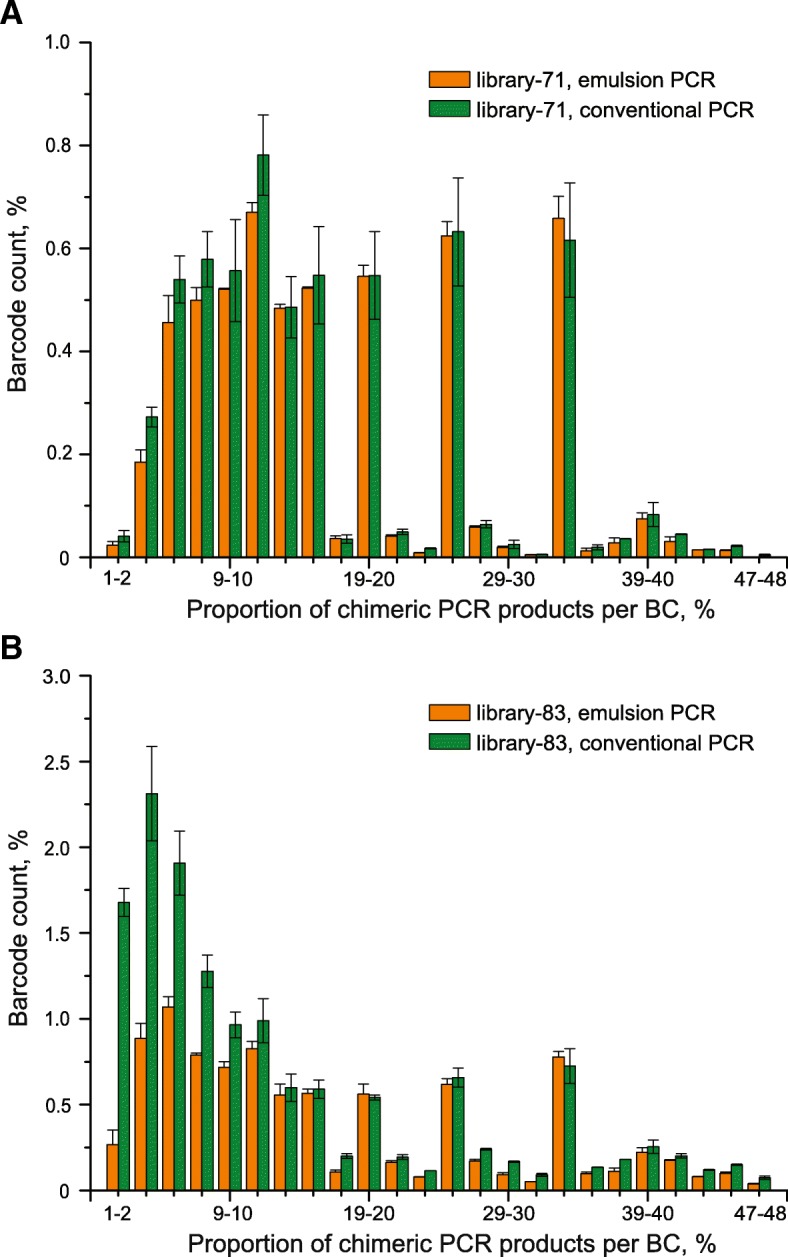
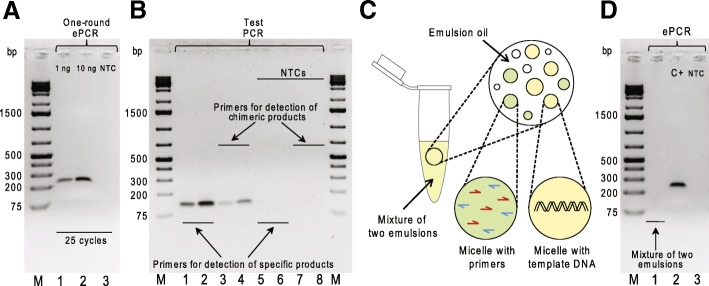
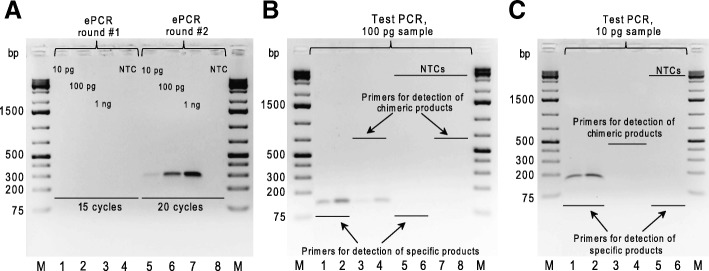
To identify settings that minimize formation of chimeric products we tested a number of PCR amplification parameters, such as conventional and emulsion types of PCR, one- or two-round amplification strategies, amount of DNA template, number of PCR cycles, and the duration of the extension step. Using specific MPRA libraries as templates, we found that the two-round amplification of the BC-ROI regions with a very low initial template amount, an elongated extension step, and a specific number of PCR cycles result in as low as 0.30 and 0.32% of chimeric products for emulsion and conventional PCR approaches, respectively.
We have identified PCR parameters that ensure synthesis of specific (non-chimeric) products from highly diverse MPRA plasmid libraries. In addition, we found that there is a negligible difference in performance of emulsion and conventional PCR approaches performed with the identified settings.
大规模平行报告分析(MPRAs)能够高通量评估各种 DNA 调控元件及其突变变体的功能。该分析基于构建高度多样化的质粒文库,文库包含两个可变片段,一个感兴趣的区域(研究序列;ROI)和一个条形码(BC),用于唯一标记每个 ROI,ROI 和 BC 由一个恒定的间隔序列隔开。文库中 BC-ROI 组合的序列可以是预先知道的,也可以是不知道的。在后一种情况下,在进行功能实验之前,必须先识别这些组合。通常,这是通过用侧翼引物扩增 BC-ROI 区域,然后对产物进行下一代测序(NGS)来完成的。然而,在扩增过程中,模板上相同间隔片段形成的嵌合 DNA 分子会严重阻碍真正的 BC-ROI 组合的识别,从而降低分析的性能。
为了确定最小化嵌合产物形成的设置,我们测试了许多 PCR 扩增参数,如常规和乳液型 PCR、一轮或两轮扩增策略、DNA 模板量、PCR 循环数和延伸步骤的持续时间。使用特定的 MPRAs 文库作为模板,我们发现,用非常低的初始模板量进行两轮 BC-ROI 区域扩增,延长延伸步骤,以及特定数量的 PCR 循环,可使乳液和常规 PCR 方法的嵌合产物分别低至 0.30%和 0.32%。
我们已经确定了确保从高度多样化的 MPRAs 质粒文库中合成特异性(非嵌合)产物的 PCR 参数。此外,我们发现,在所确定的条件下,乳液和常规 PCR 方法的性能差异可以忽略不计。