Institute of Pharmaceutical Chemistry, Goethe University Frankfurt, Frankfurt, Germany.
Institute of Pharmaceutical Chemistry, Goethe University Frankfurt, Frankfurt, Germany; Assay development and screening, Fraunhofer Institute for Translational Medicine and Pharmacology ITMP, Frankfurt, Germany.
J Biol Chem. 2021 Jul;297(1):100814. doi: 10.1016/j.jbc.2021.100814. Epub 2021 May 31.
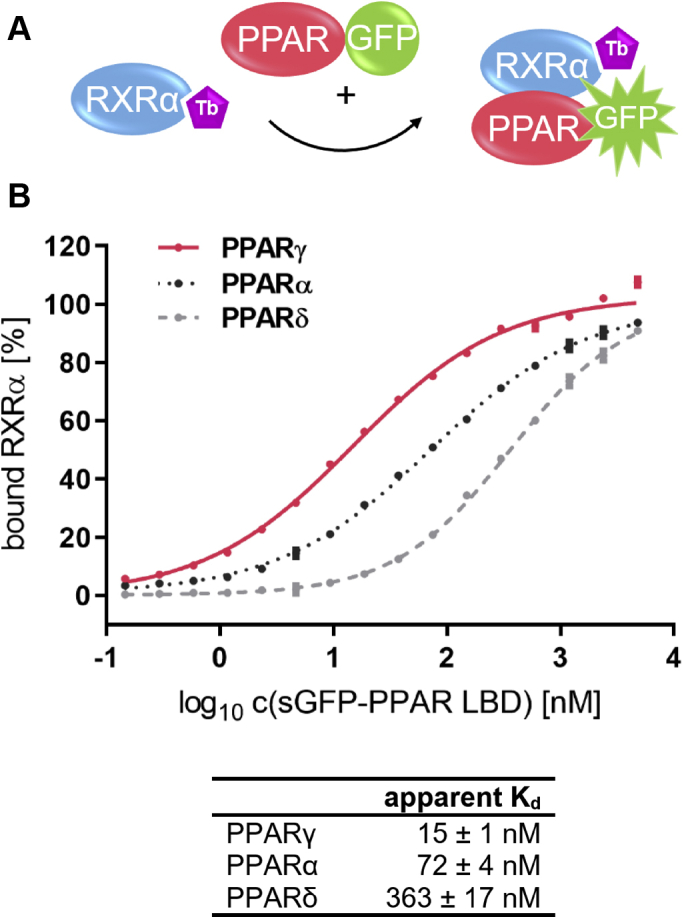
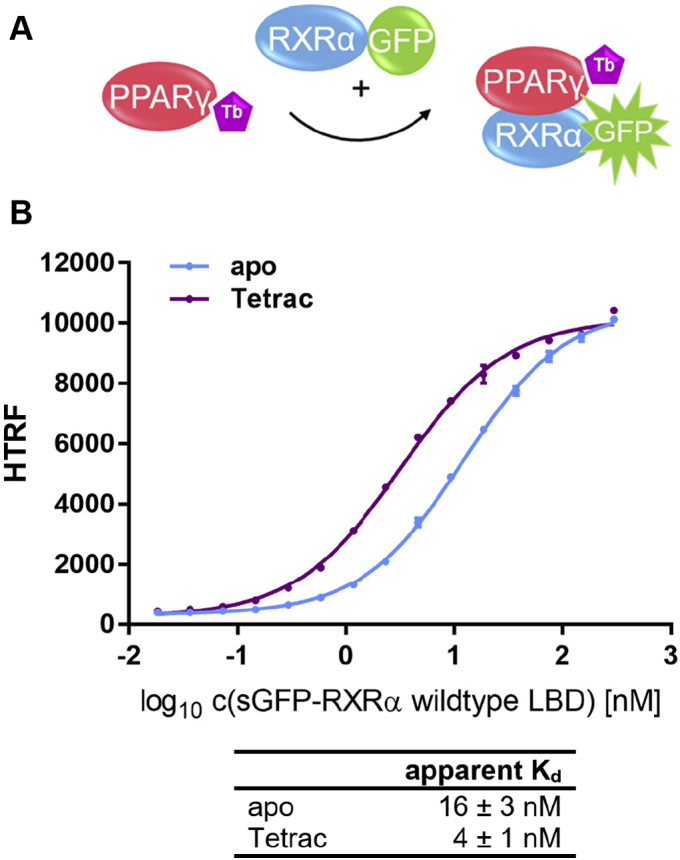
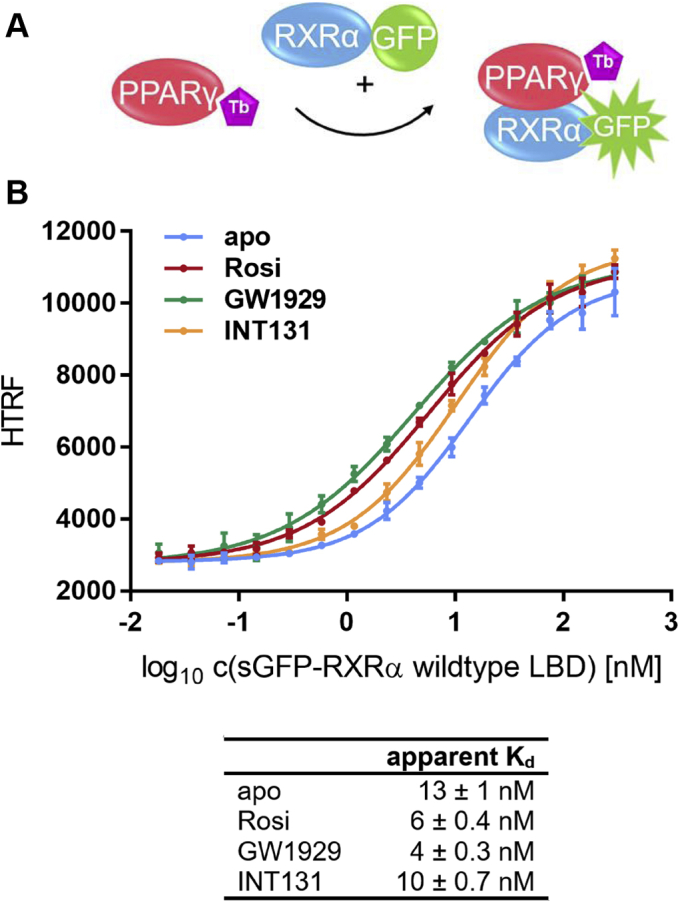
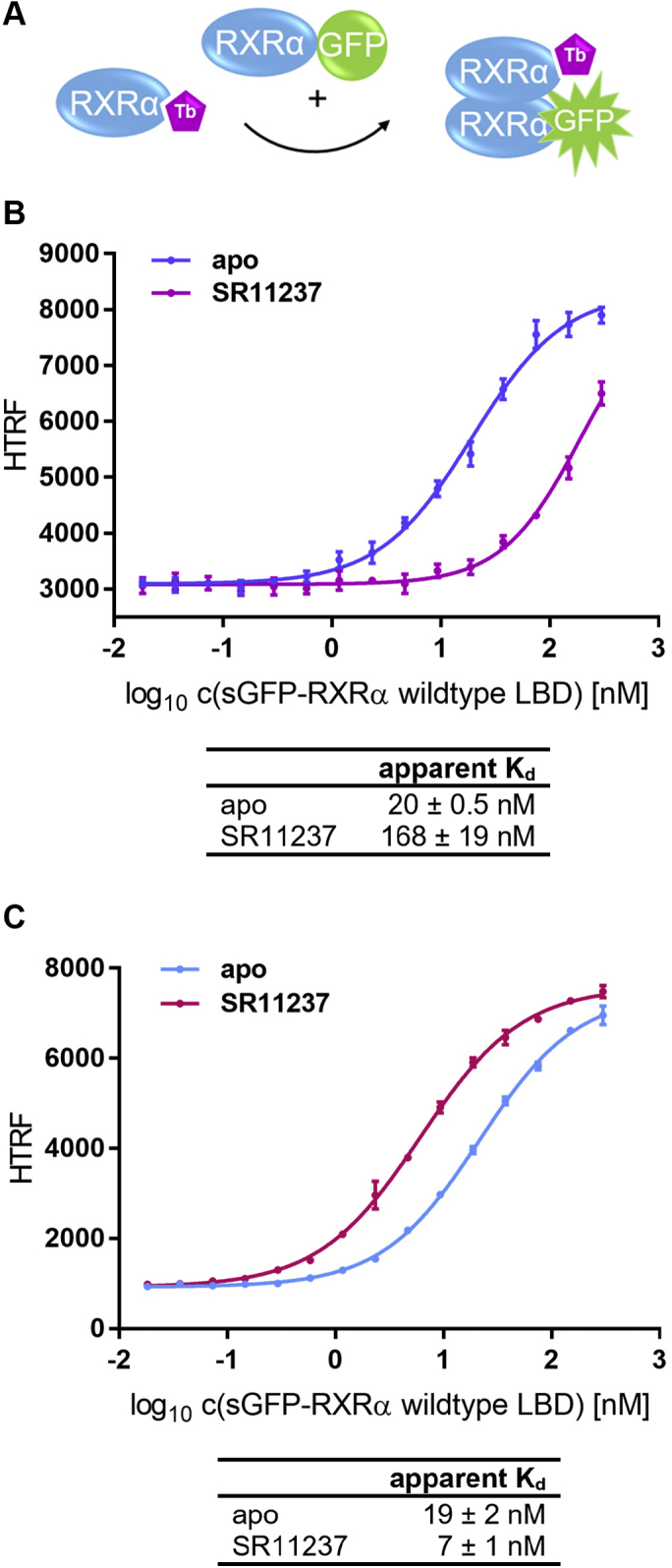
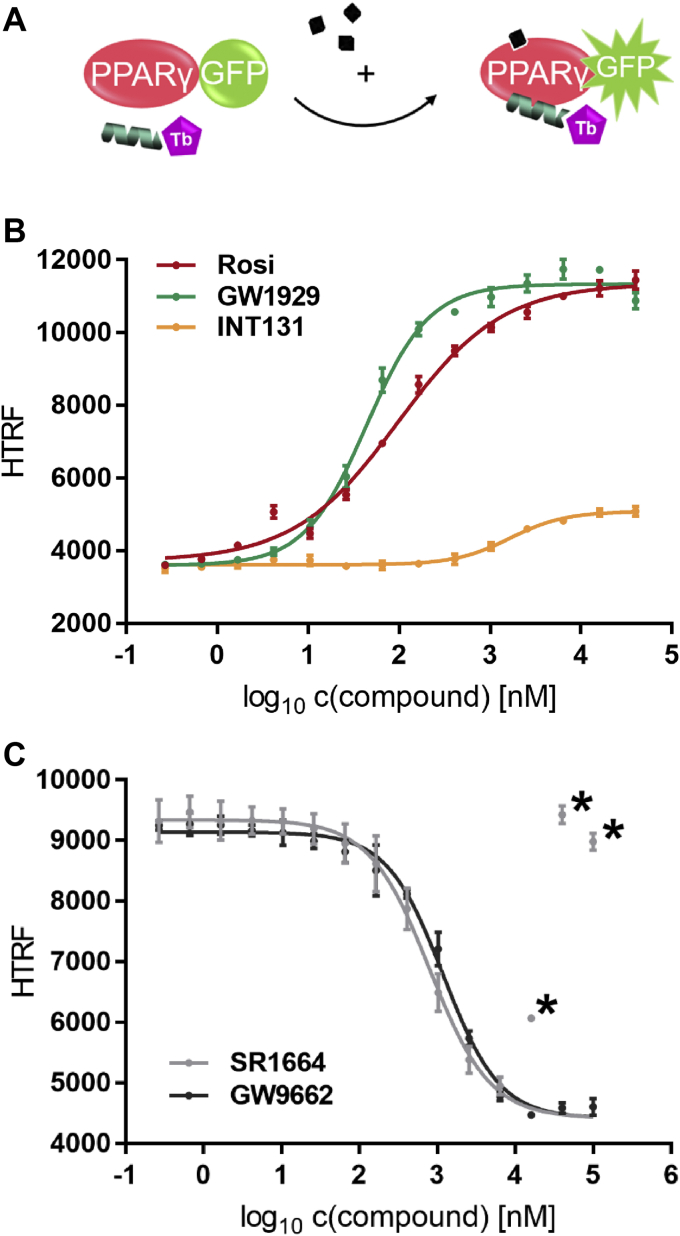
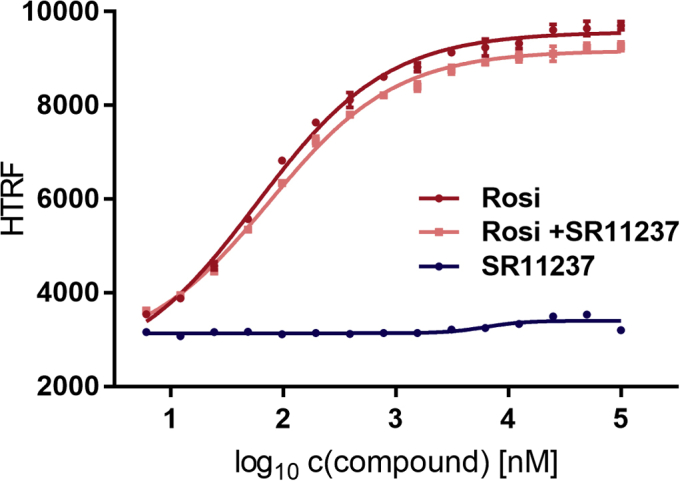
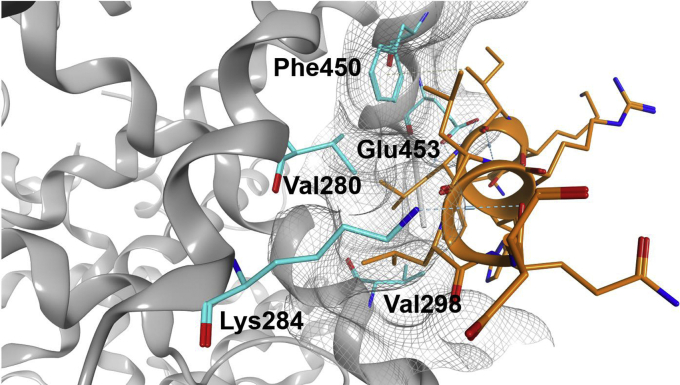
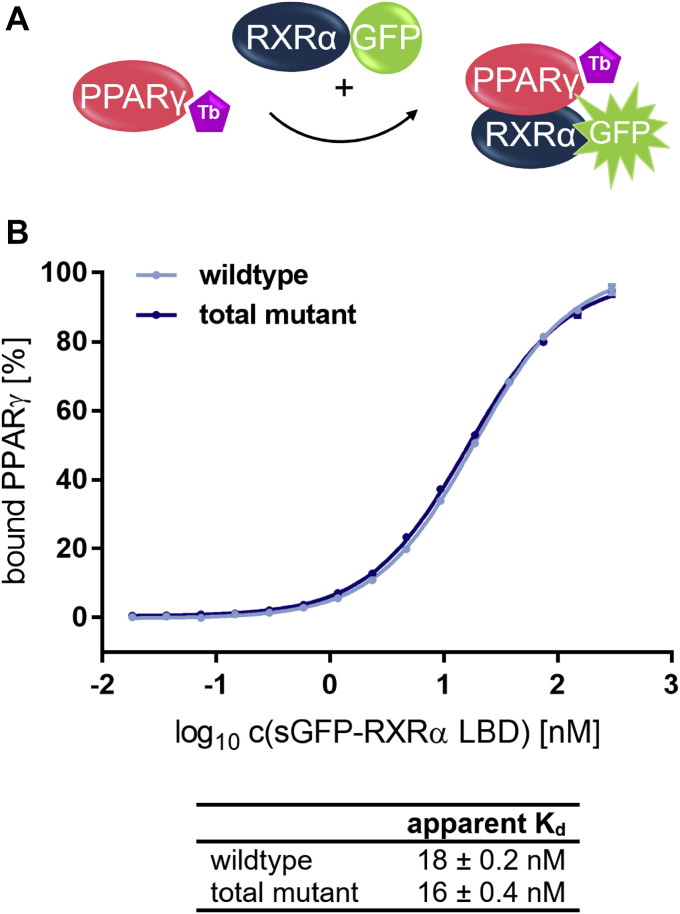
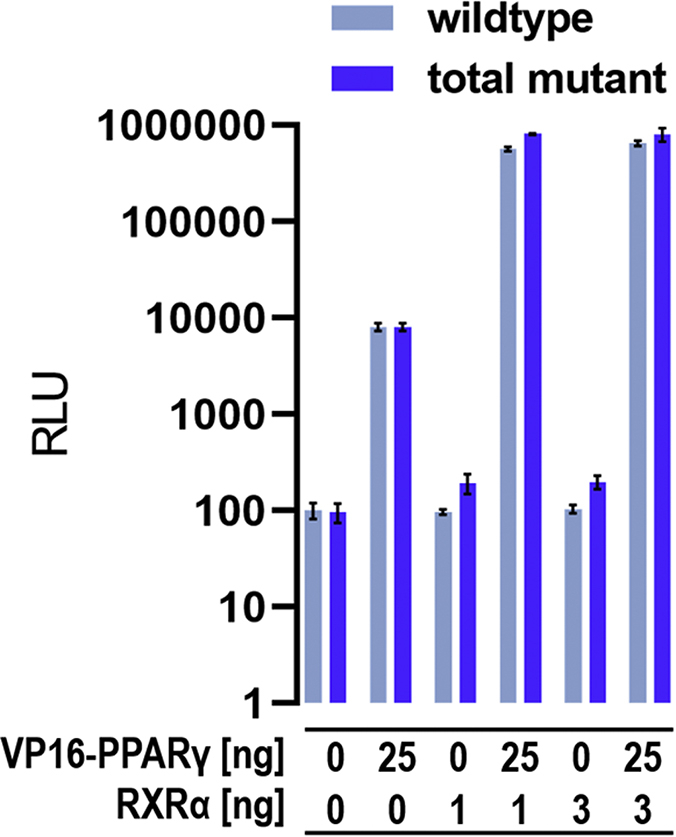
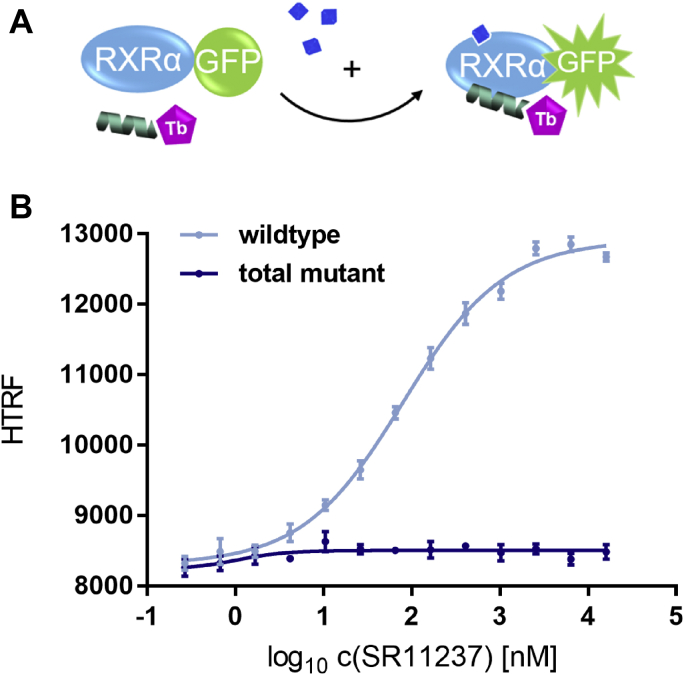
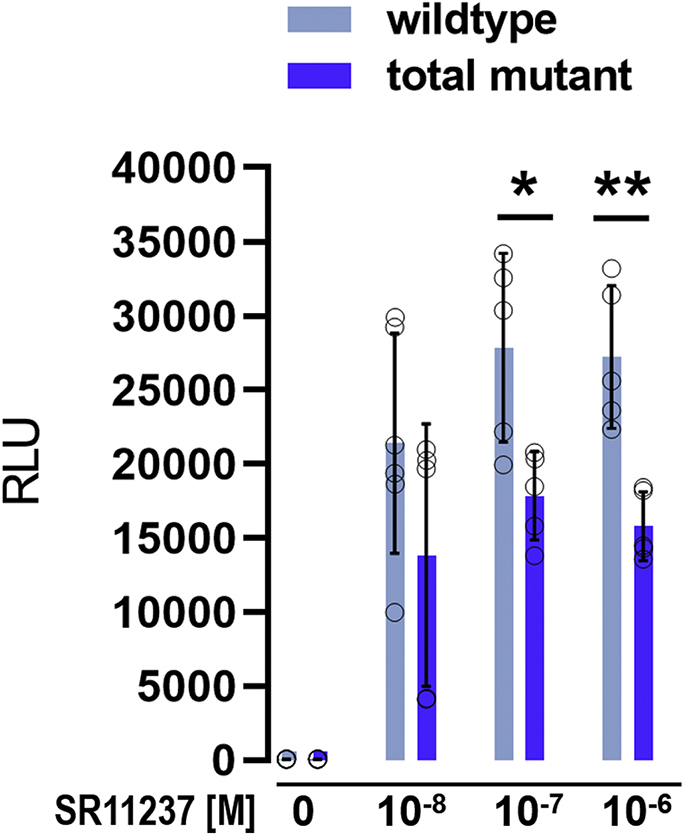
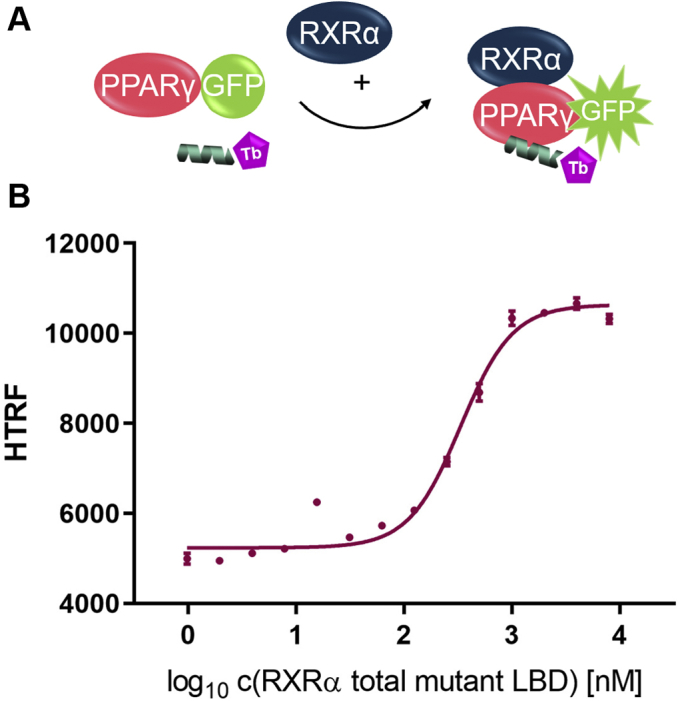
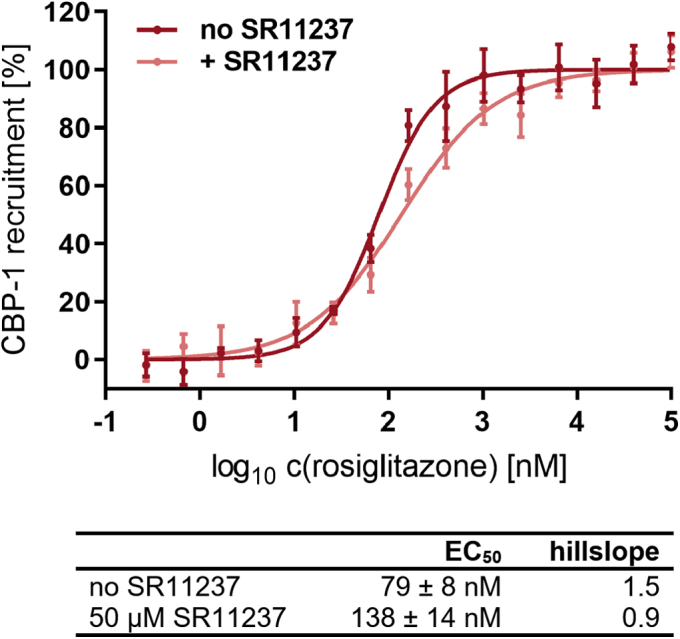
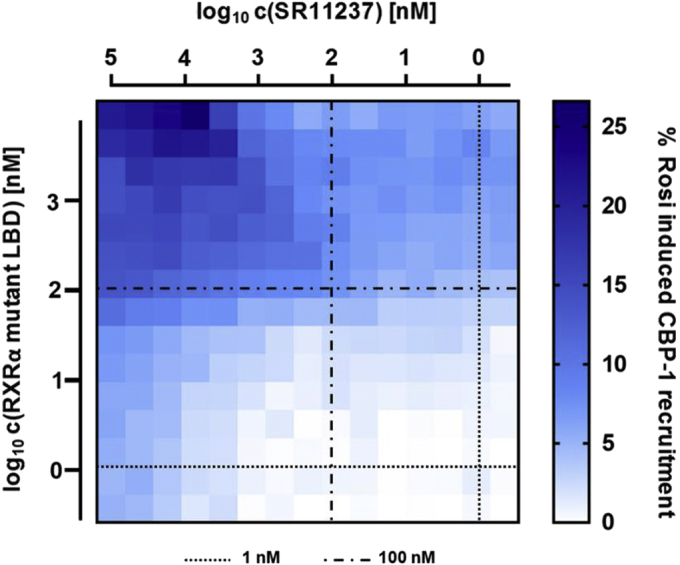
Nuclear receptors (NRs) activate transcription of target genes in response to binding of ligands to their ligand-binding domains (LBDs). Typically, in vitro assays use either gene expression or the recruitment of coactivators to the isolated LBD of the NR of interest to measure NR activation. However, this approach ignores that NRs function as homo- as well as heterodimers and that the LBD harbors the main dimerization interface. Cofactor recruitment is thereby interconnected with oligomerization status as well as ligand occupation of the partnering LBD through allosteric cross talk. Here we present a modular set of homogeneous time-resolved FRET-based assays through which we investigated the activation of PPARγ in response to ligands and the formation of heterodimers with its obligatory partner RXRα. We introduced mutations into the RXRα LBD that prevent coactivator binding but do not interfere with LBD dimerization or ligand binding. This enabled us to specifically detect PPARγ coactivator recruitment to PPARγ:RXRα heterodimers. We found that the RXRα agonist SR11237 destabilized the RXRα homodimer but promoted formation of the PPARγ:RXRα heterodimer, while being inactive on PPARγ itself. Of interest, incorporation of PPARγ into the heterodimer resulted in a substantial gain in affinity for coactivator CBP-1, even in the absence of ligands. Consequently, SR11237 indirectly promoted coactivator binding to PPARγ by shifting the oligomerization preference of RXRα toward PPARγ:RXRα heterodimer formation. These results emphasize that investigation of ligand-dependent NR activation should take NR dimerization into account. We envision these assays as the necessary assay tool kit for investigating NRs that partner with RXRα.
核受体 (NRs) 通过与配体结合到其配体结合域 (LBD) 来激活靶基因的转录。通常,体外测定使用基因表达或募集共激活因子到感兴趣的 NR 的分离 LBD 来测量 NR 激活。然而,这种方法忽略了 NR 作为同源二聚体和异源二聚体起作用,并且 LBD 包含主要的二聚化界面。因此,共激活因子的募集与寡聚化状态以及配体对伴侣 LBD 的占据通过变构串扰相互关联。在这里,我们提出了一组模块化的均相时间分辨荧光共振能量转移 (FRET) 测定法,通过这些方法,我们研究了 PPARγ 对配体的激活以及与其必需伴侣 RXRα 形成异源二聚体的情况。我们引入了突变到 RXRα LBD 中,这些突变阻止了共激活因子的结合,但不干扰 LBD 二聚化或配体结合。这使我们能够特异性地检测到 PPARγ 共激活因子募集到 PPARγ:RXRα 异源二聚体。我们发现,RXRα 激动剂 SR11237 破坏了 RXRα 同源二聚体的稳定性,但促进了 PPARγ:RXRα 异源二聚体的形成,而对 PPARγ 本身没有活性。有趣的是,即使在没有配体的情况下,将 PPARγ 纳入异源二聚体也导致对共激活因子 CBP-1 的亲和力大大提高。因此,SR11237 通过将 RXRα 的寡聚化偏好转向 PPARγ:RXRα 异源二聚体形成,间接促进了共激活因子与 PPARγ 的结合。这些结果强调了研究配体依赖性 NR 激活应该考虑 NR 二聚化。我们设想这些测定法是研究与 RXRα 结合的 NR 的必要测定工具包。