, Roboleo & Co, Leeds, UK.
Orphanet J Rare Dis. 2021 Jun 3;16(1):255. doi: 10.1186/s13023-021-01884-4.
Progressive familial intrahepatic cholestasis is a rare, heterogeneous group of liver disorders of autosomal recessive inheritance, characterised by an early onset of cholestasis with pruritus and malabsorption, which rapidly progresses, eventually culminating in liver failure. For children and their parents, PFIC is an extremely distressing disease. Significant pruritus can lead to severe cutaneous mutilation and may affect many activities of daily living through loss of sleep, irritability, poor attention, and impaired school performance.
Databases including MEDLINE and Embase were searched for publications on PFIC prevalence, incidence or natural history, and the economic burden or health-related quality of life of patients with PFIC. Preferred Reporting Items for Systematic Reviews and Meta-Analyses guidelines were followed.
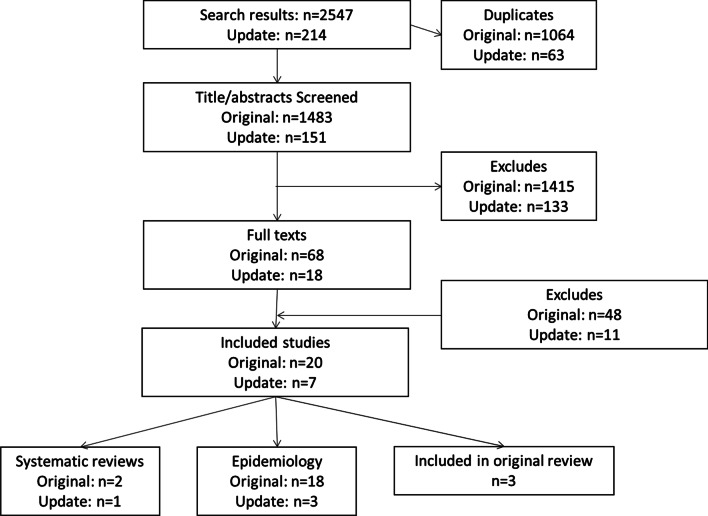
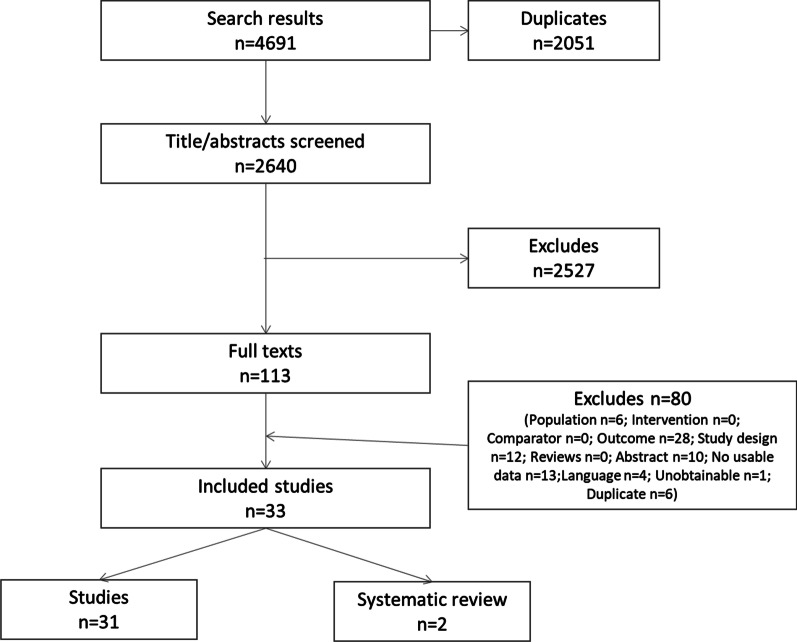
Three systematic reviews and twenty-two studies were eligible for inclusion for the epidemiology of PFIC including a total of 2603 patients. Study periods ranged from 3 to 33 years. Local population prevalence of PFIC was reported in three studies, ranging from 9.0 to 12.0% of children admitted with cholestasis, acute liver failure, or splenomegaly. The most detailed data come from the NAPPED study where native liver survival of >15 years is predicted in PFIC2 patients with a serum bile acid concentration below 102 µmol/L following bile diversion surgery. Burden of disease was mainly reported through health-related quality of life (HRQL), rates of surgery and survival. Rates of biliary diversion and liver transplant varied widely depending on study period, sample size and PFIC type, with many patients have multiple surgeries and progressing to liver transplant. This renders data unsuitable for comparison.
Using robust and transparent methods, this systematic review summarises our current knowledge of PFIC. The epidemiological overview is highly mixed and dependent on presentation and PFIC subtype. Only two studies reported HRQL and mortality results were variable across different subtypes. Lack of data and extensive heterogeneity severely limit understanding across this disease area, particularly variation around and within subtypes.
进行性家族性肝内胆汁淤积症是一种罕见的、异质性的常染色体隐性遗传性肝脏疾病,其特征为早发性胆汁淤积伴瘙痒和吸收不良,且病情迅速进展,最终导致肝功能衰竭。对于儿童及其父母来说,PFIC 是一种极其痛苦的疾病。严重的瘙痒可导致严重的皮肤损伤,并可通过睡眠不足、烦躁不安、注意力不集中和学习成绩下降等方式影响许多日常生活活动。
检索 MEDLINE 和 Embase 等数据库,以获取关于 PFIC 患病率、发病率或自然史,以及 PFIC 患者的经济负担或健康相关生活质量的研究报告。研究遵循系统评价和荟萃分析的 Preferred Reporting Items 指南。
纳入了 3 项系统评价和 22 项研究,这些研究涉及 PFIC 的流行病学,共纳入 2603 例患者。研究期间从 3 年到 33 年不等。有 3 项研究报道了当地人群中 PFIC 的患病率,范围为因胆汁淤积、急性肝衰竭或脾肿大而住院的儿童的 9.0%至 12.0%。最详细的数据来自 NAPPED 研究,该研究预测血清胆汁酸浓度低于 102μmol/L 的 PFIC2 患者在进行胆汁引流手术后,超过 15 年的原发性肝存活率。疾病负担主要通过健康相关生活质量(HRQL)、手术率和生存率来报告。胆道分流术和肝移植的比例因研究期间、样本量和 PFIC 类型而异,许多患者需要多次手术,最终进展为肝移植。这使得数据不适合比较。
本系统评价采用了可靠和透明的方法,总结了我们目前对 PFIC 的认识。流行病学概述高度混杂,取决于临床表现和 PFIC 亚型。只有两项研究报告了 HRQL 和死亡率结果,且在不同亚型之间存在差异。缺乏数据和广泛的异质性严重限制了对该病的理解,尤其是在不同亚型之间和内部的差异。