Complex Systems Division, Beijing Computational Science Research Center, Haidian District, Beijing, 100193, China.
Complex Systems Division, Beijing Computational Science Research Center, Haidian District, Beijing, 100193, China.
Comput Biol Med. 2021 Aug;135:104634. doi: 10.1016/j.compbiomed.2021.104634. Epub 2021 Jul 6.
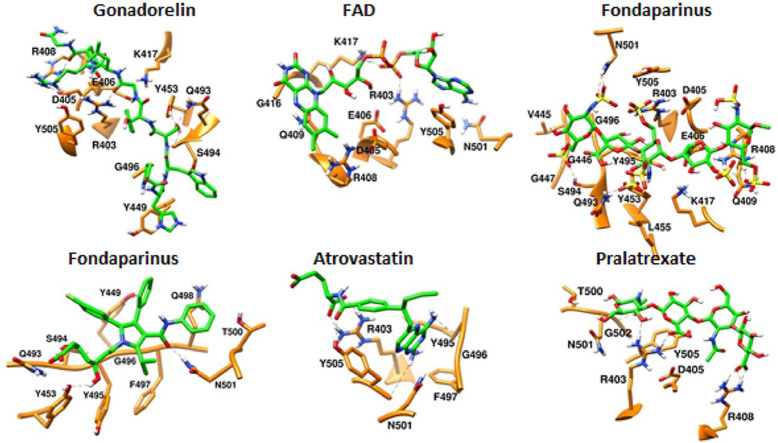
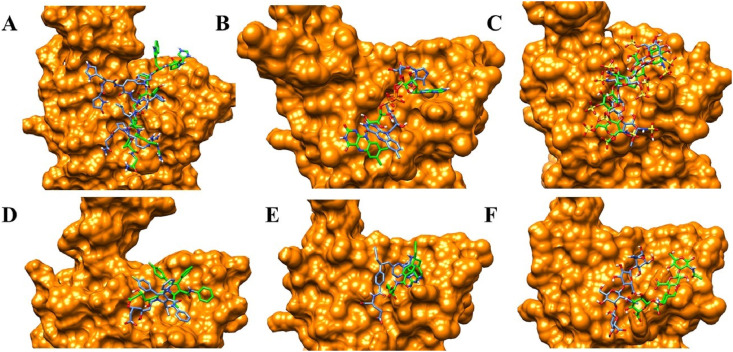
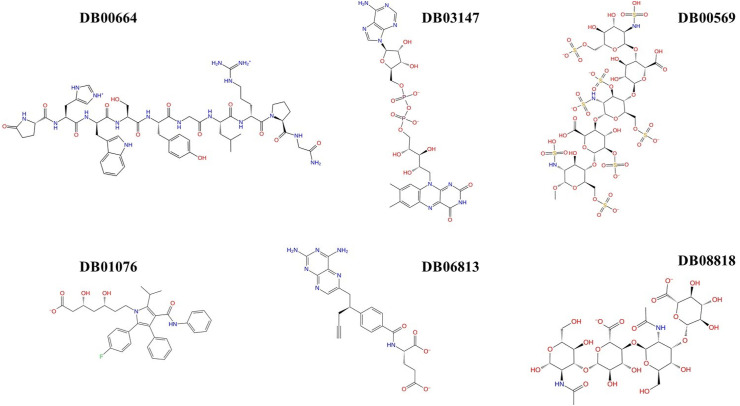
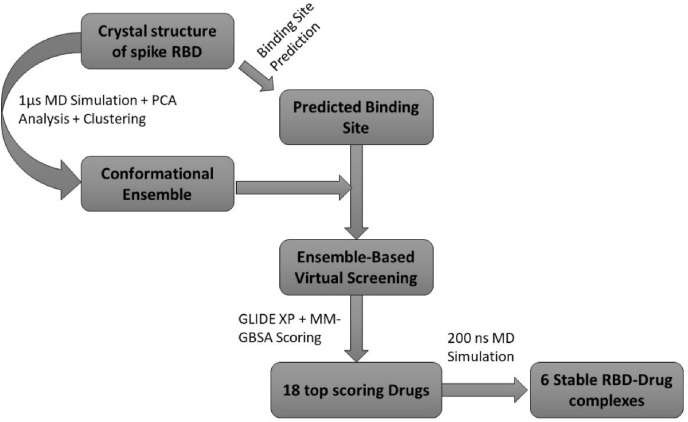
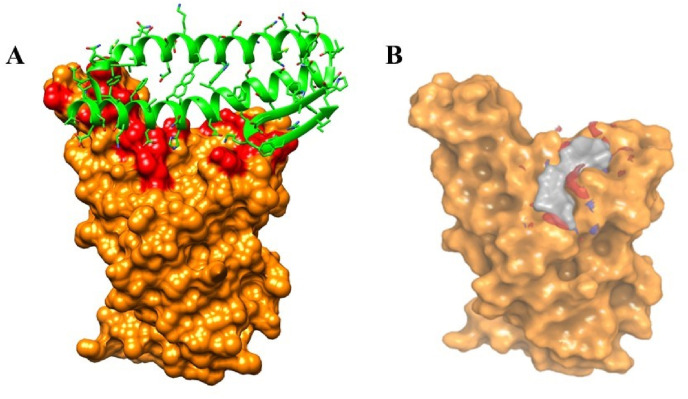
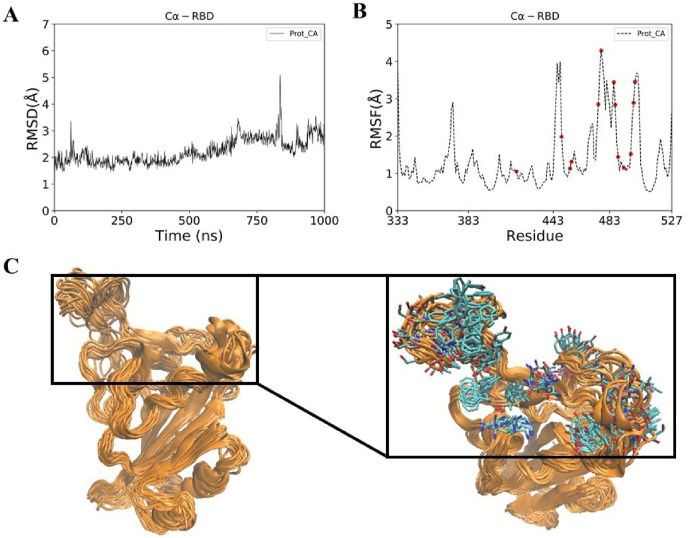
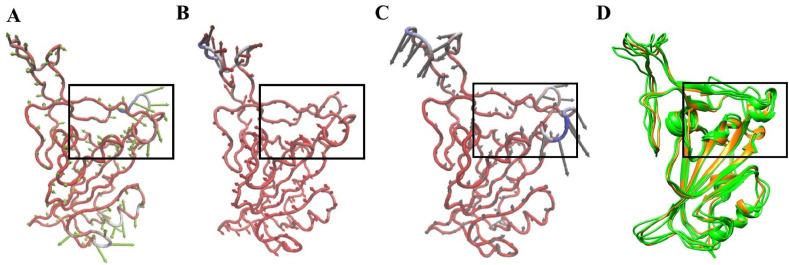
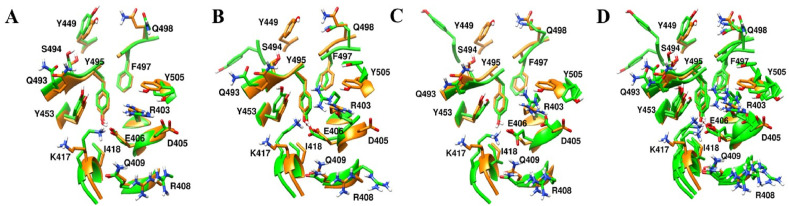
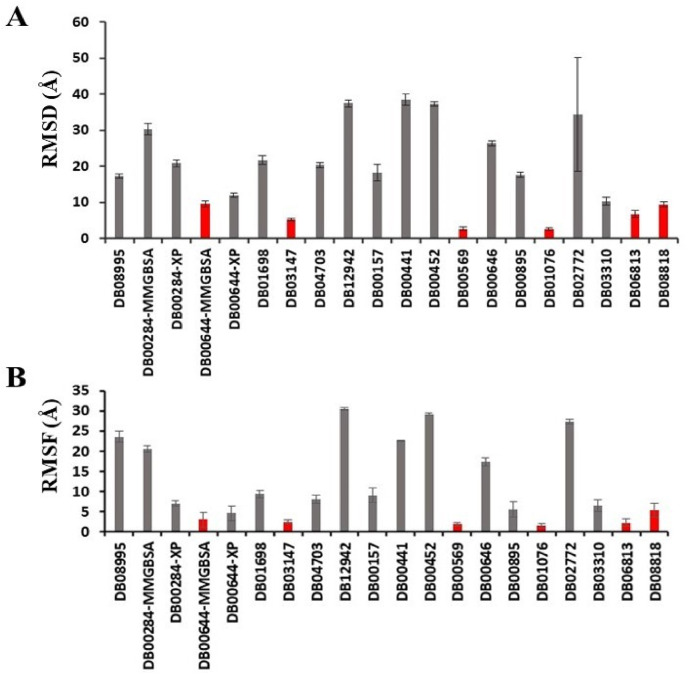
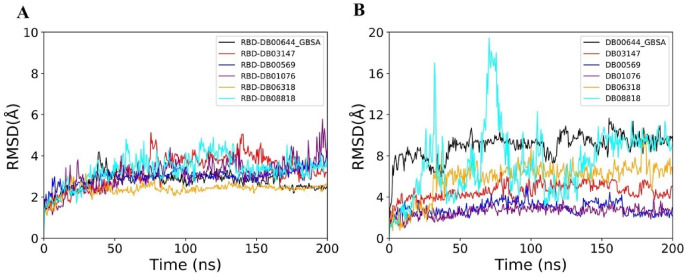
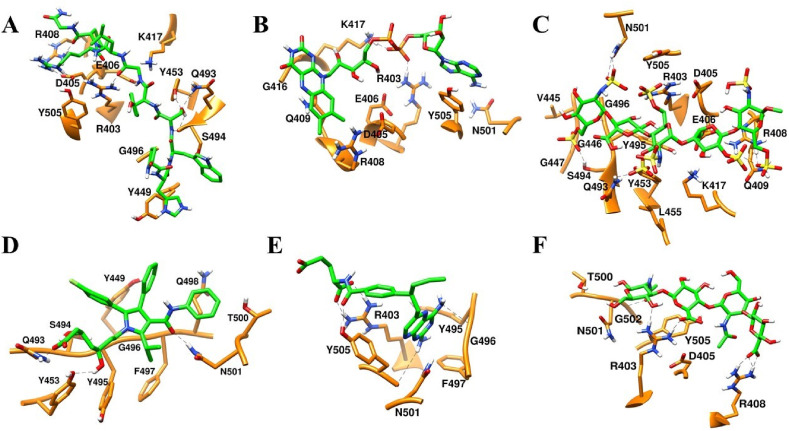
Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has caused worldwide pandemic and is responsible for millions of worldwide deaths due to -a respiratory disease known as COVID-19. In the search for a cure of COVID-19, drug repurposing is a fast and cost-effective approach to identify anti-COVID-19 drugs from existing drugs. The receptor binding domain (RBD) of the SARS-CoV-2 spike protein has been a main target for drug designs to block spike protein binding to ACE2 proteins. In this study, we probed the conformational plasticity of the RBD using long molecular dynamics (MD) simulations, from which, representative conformations were identified using clustering analysis. Three simulated conformations and the original crystal structure were used to screen FDA approved drugs (2466 drugs) against the predicted binding site at the ACE2-RBD interface, leading to 18 drugs with top docking scores. Notably, 16 out of the 18 drugs were obtained from the simulated conformations, while the crystal structure suggests poor binding. The binding stability of the 18 drugs were further investigated using MD simulations. Encouragingly, 6 drugs exhibited stable binding with RBD at the ACE2-RBD interface and 3 of them (gonadorelin, fondaparinux and atorvastatin) showed significantly enhanced binding after the MD simulations. Our study shows that flexibility modeling of SARS-CoV-2 RBD using MD simulation is of great help in identifying novel agents which might block the interaction between human ACE2 and the SARS-CoV-2 RBD for inhibiting the virus infection.
严重急性呼吸综合征冠状病毒 2(SARS-CoV-2)已在全球范围内引发大流行,并导致数以百万计的人因被称为 COVID-19 的呼吸道疾病而死亡。在寻找 COVID-19 的治疗方法时,药物再利用是一种快速且具有成本效益的方法,可以从现有药物中识别出抗 COVID-19 的药物。SARS-CoV-2 刺突蛋白的受体结合结构域(RBD)一直是药物设计的主要目标,旨在阻止刺突蛋白与 ACE2 蛋白结合。在这项研究中,我们使用长分子动力学(MD)模拟来探测 RBD 的构象可塑性,通过聚类分析确定了代表性构象。使用三种模拟构象和原始晶体结构来筛选美国食品和药物管理局批准的药物(2466 种药物)针对 ACE2-RBD 界面上预测的结合位点,得到了 18 种对接得分最高的药物。值得注意的是,这 18 种药物中有 16 种是从模拟构象中获得的,而晶体结构则表明结合较差。进一步使用 MD 模拟研究了 18 种药物的结合稳定性。令人鼓舞的是,6 种药物在 ACE2-RBD 界面与 RBD 表现出稳定的结合,其中 3 种(促性腺激素释放激素、磺达肝癸钠和阿托伐他汀)在 MD 模拟后表现出显著增强的结合。我们的研究表明,使用 MD 模拟对 SARS-CoV-2 RBD 进行柔性建模有助于识别可能阻断人类 ACE2 与 SARS-CoV-2 RBD 相互作用的新型药物,从而抑制病毒感染。