Manchester Institute of Biotechnology, The University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK.
Department of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK.
J Comput Aided Mol Des. 2021 Aug;35(8):911-921. doi: 10.1007/s10822-021-00406-5. Epub 2021 Jul 15.
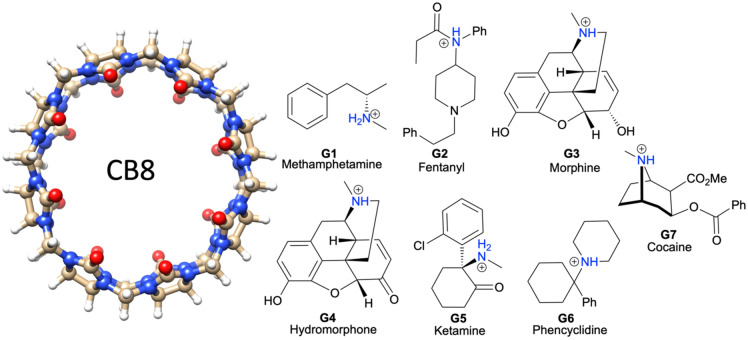
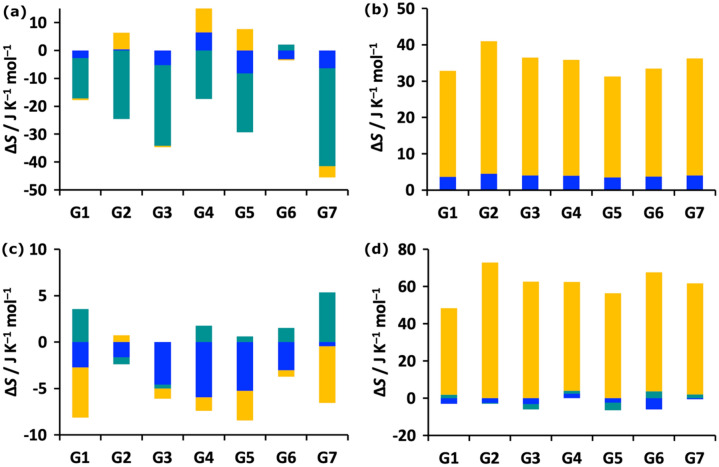
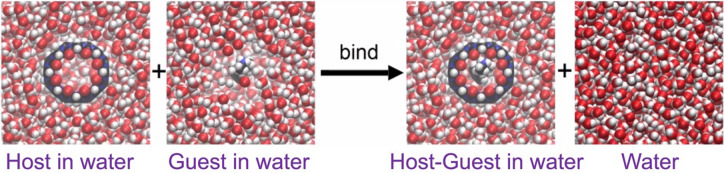
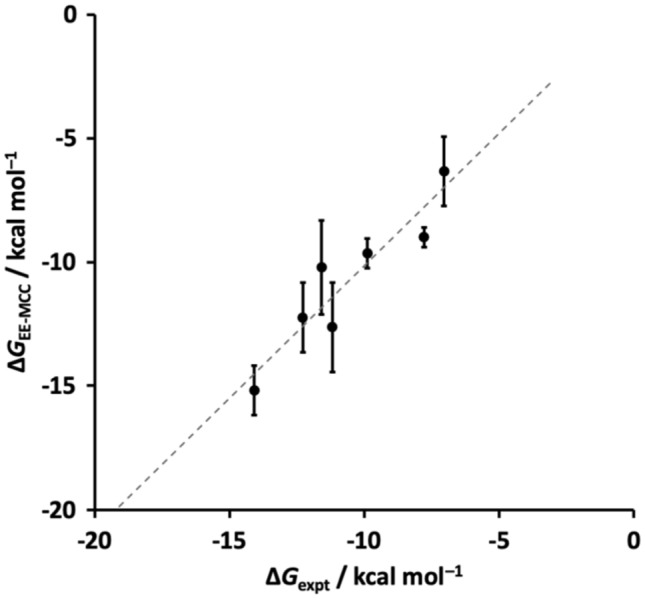
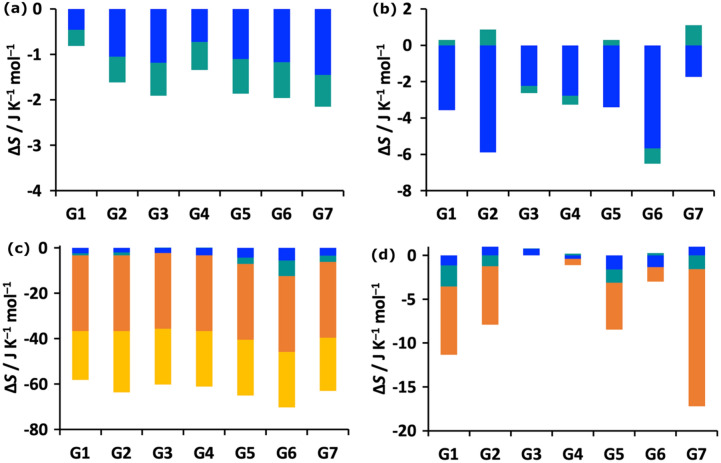
Free energy drives a wide range of molecular processes such as solvation, binding, chemical reactions and conformational change. Given the central importance of binding, a wide range of methods exist to calculate it, whether based on scoring functions, machine-learning, classical or electronic structure methods, alchemy, or explicit evaluation of energy and entropy. Here we present a new energy-entropy (EE) method to calculate the host-guest binding free energy directly from molecular dynamics (MD) simulation. Entropy is evaluated using Multiscale Cell Correlation (MCC) which uses force and torque covariance and contacts at two different length scales. The method is tested on a series of seven host-guest complexes in the SAMPL8 (Statistical Assessment of the Modeling of Proteins and Ligands) "Drugs of Abuse" Blind Challenge. The EE-MCC binding free energies are found to agree with experiment with an average error of 0.9 kcal mol. MCC makes clear the origin of the entropy changes, showing that the large loss of positional, orientational, and to a lesser extent conformational entropy of each binding guest is compensated for by a gain in orientational entropy of water released to bulk, combined with smaller decreases in vibrational entropy of the host, guest and contacting water.
自由能驱动着广泛的分子过程,如溶剂化、结合、化学反应和构象变化。鉴于结合的重要性,存在广泛的方法来计算它,无论是基于评分函数、机器学习、经典或电子结构方法、炼金术,还是对能量和熵的显式评估。在这里,我们提出了一种新的能量-熵 (EE) 方法,可直接从分子动力学 (MD) 模拟中计算主客体结合自由能。熵是使用 Multiscale Cell Correlation (MCC) 评估的,该方法使用力和扭矩协方差以及两个不同长度尺度的接触。该方法在 SAMPL8(蛋白质和配体建模的统计评估)“滥用药物”盲测中的七个主客体复合物系列上进行了测试。EE-MCC 结合自由能与实验结果一致,平均误差为 0.9 kcal/mol。MCC 清楚地显示了熵变化的起源,表明每个结合客体的位置、取向和在较小程度上构象熵的大量损失被释放到主体的水的取向熵的增加所补偿,同时宿主、客体和接触水的振动熵也略有减少。