Department of Pediatrics and Pediatric Endocrinology, Faculty of Medical Sciences in Katowice, Medical University of Silesia, 40-752 Katowice, Poland.
Department of Genetics, Polish Mother's Memorial Hospital Research Institute, 93-338 Lodz, Poland.
Int J Environ Res Public Health. 2021 Jul 5;18(13):7186. doi: 10.3390/ijerph18137186.
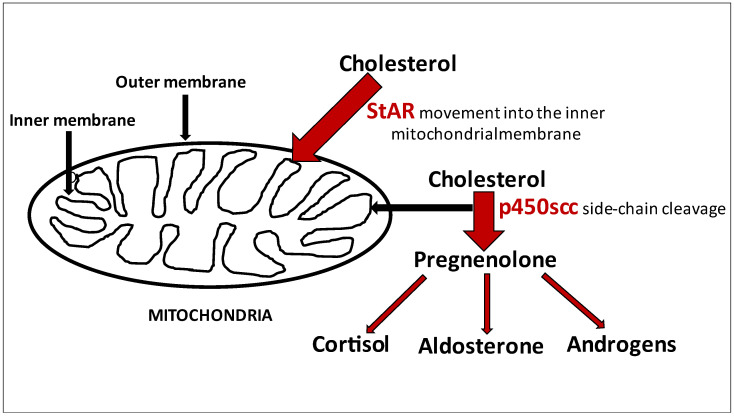
A novel : c.1236 + 5G > A was identified, expanding the mutation spectrum of the congenital adrenal insufficiency with 46,XY sex reversal. In a now 17-year-old girl delivered full-term (G2P2, parents unrelated), adrenal failure was diagnosed in the first year of life based on clinical picture of acute adrenal crisis with vomiting, dehydration, weight loss, hypotension, and electrolyte disturbances. At the time, hormonal tests revealed primary adrenocortical insufficiency and steroid profiles showed lack of products of steroidogenesis, and since then the patient has been treated with substitution doses of hydrocortisone and fludrocortisone. At the age of 14, considering the absence of puberty symptoms, extended diagnostic tests revealed elevated LH levels (26.5 mIU/mL) with pre-puberty FSH levels (4.9 mIU/mL), low estradiol (28 pmol/L), testosterone (<2.5 ng/mL), and extremely high levels of ACTH (4961 pg/mL). A cytogenetic study revealed a 46 XY karyotype. A molecular examination confirmed the missense mutation and a novel splice-site mutation of gene. Compound heterozygosity for the gene with a known pathogenic variant in one allele and a novel splice site mutation in the second allele is most probably responsible for congenital adrenal insufficiency with 46,XY sex reversal. We discuss the necessity of cytogenetic test in the case of early onset of adrenal failure in the absence of steroidogenesis metabolites in the steroid profile.
c.1236 + 5G > A 被发现,扩大了先天性肾上腺皮质功能减退症伴 46,XY 性别反转的突变谱。在一个现在 17 岁的足月(G2P2,父母无血缘关系)出生的女孩中,根据急性肾上腺危象的临床表现,即呕吐、脱水、体重减轻、低血压和电解质紊乱,在出生后的第一年就诊断出肾上腺功能衰竭。当时,激素检测显示原发性肾上腺皮质功能不全,类固醇谱显示缺乏类固醇生成产物,此后患者一直接受氢化可的松和氟氢可的松替代剂量治疗。14 岁时,由于没有青春期症状,延长的诊断测试显示 LH 水平升高(26.5 mIU/mL),同时伴有青春期前 FSH 水平(4.9 mIU/mL)、低雌二醇(28 pmol/L)、睾酮(<2.5 ng/mL)和极高的 ACTH 水平(4961 pg/mL)。细胞遗传学研究显示 46 XY 核型。分子检查证实了 基因的错义突变和新的剪接位点突变。基因的复合杂合性,一个等位基因携带已知的致病性变异,另一个等位基因携带新的剪接位点突变,很可能是导致先天性肾上腺皮质功能减退症伴 46,XY 性别反转的原因。我们讨论了在类固醇谱中缺乏类固醇生成代谢物的情况下,出现早期肾上腺功能衰竭时进行细胞遗传学检查的必要性。