Mateo-Estrada Valeria, Fernández-Vázquez José Luis, Moreno-Manjón Julia, Hernández-González Ismael L, Rodríguez-Noriega Eduardo, Morfín-Otero Rayo, Alcántar-Curiel María Dolores, Castillo-Ramírez Santiago
Programa de Genómica Evolutiva, Centro de Ciencias Genómicas, Universidad Nacional Autónoma de México, Cuernavaca, México.
Laboratorio de Infectología, Microbiología e Inmunología Clínica, Unidad de Investigación en Medicina Experimental, Facultad de Medicina, Universidad Nacional Autónoma de México, Ciudad de México, México.
mSystems. 2021 Aug 31;6(4):e0062621. doi: 10.1128/mSystems.00626-21. Epub 2021 Jul 20.
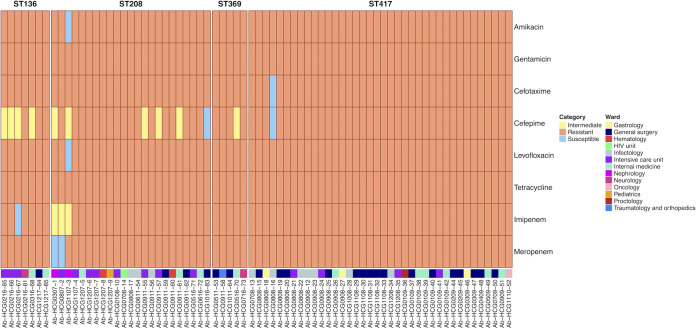
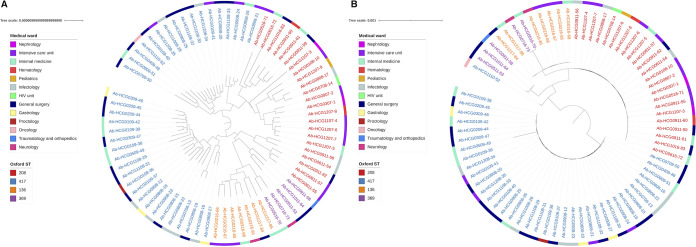
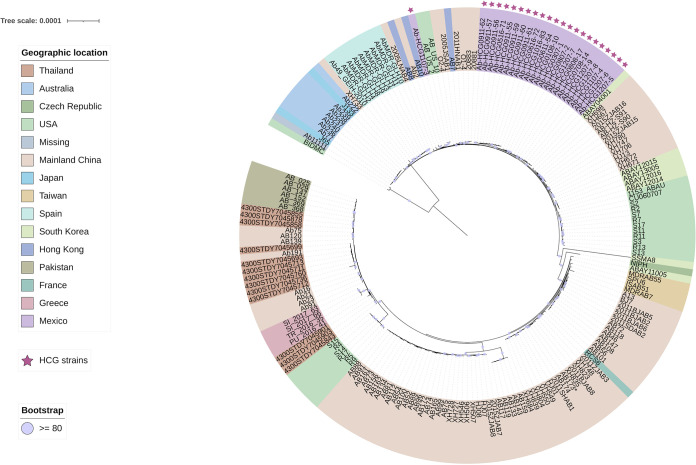
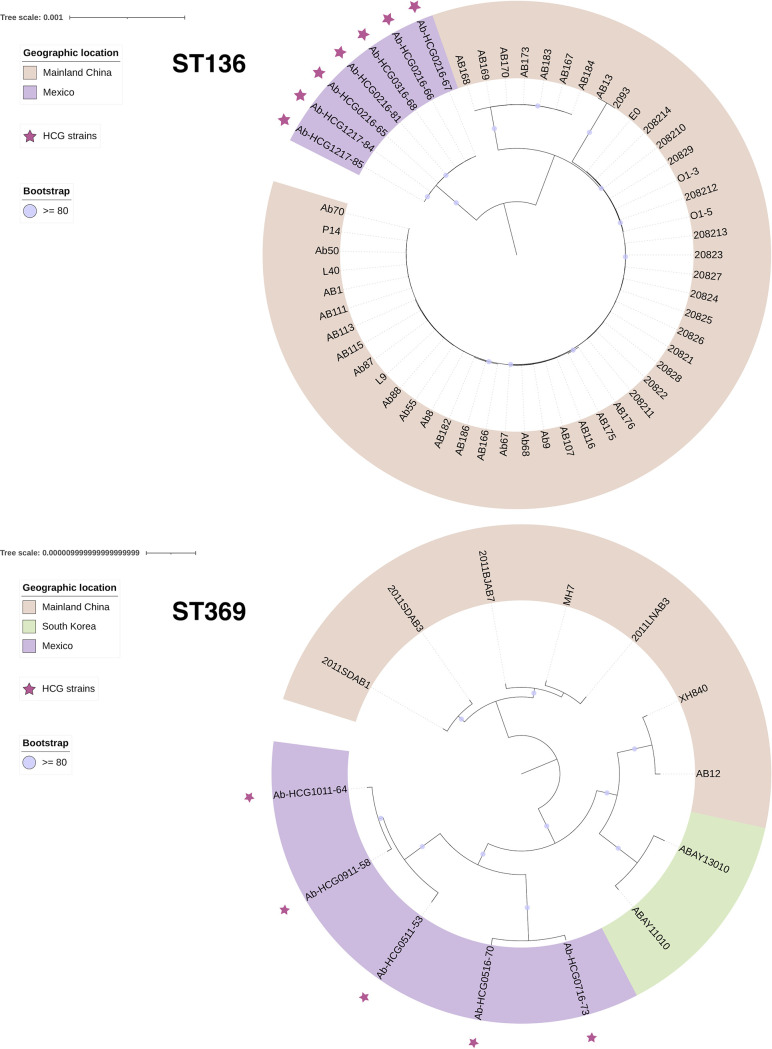
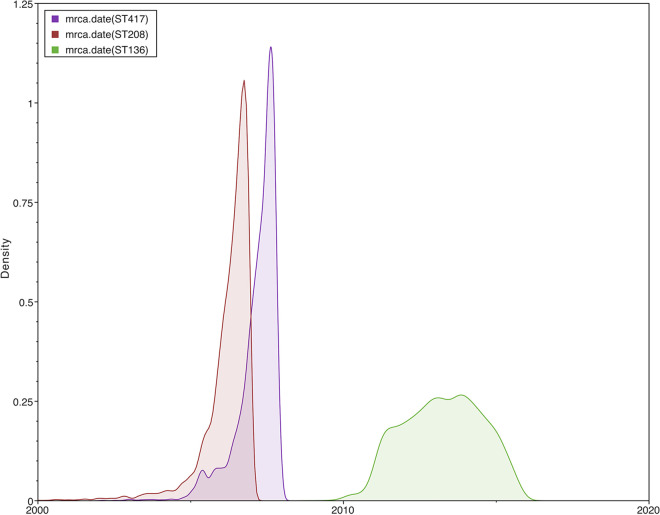
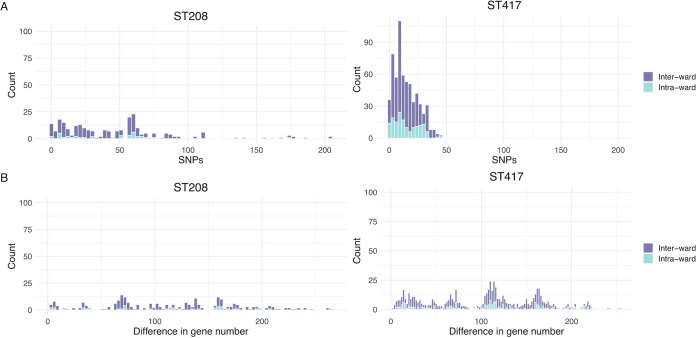
Acinetobacter baumannii has become one of the most important multidrug-resistant nosocomial pathogens all over the world. Nonetheless, very little is known about the diversity of A. baumannii lineages coexisting in hospital settings. Here, using whole-genome sequencing, epidemiological data, and antimicrobial susceptibility tests, we uncover the transmission dynamics of extensive and multidrug-resistant A. baumannii in a tertiary hospital over a decade. Our core genome phylogeny of almost 300 genomes suggests that there were several introductions of lineages from international clone 2 into the hospital. The molecular dating analysis shows that these introductions happened in 2006, 2007, and 2013. Furthermore, using the accessory genome, we show that these lineages were extensively disseminated across many wards in the hospital. Our results demonstrate that accessory genome variation can be a very powerful tool for conducting genomic epidemiology. We anticipate future studies employing the accessory genome along with the core genome as a powerful phylogenomic strategy to track bacterial transmissions over very short microevolutionary scales. Whole-genome sequencing for epidemiological investigations (genomic epidemiology) has been of paramount importance to understand the transmission dynamics of many bacterial (and nonbacterial) pathogens. Commonly, variation in the core genome, single nucleotide polymorphisms (SNPs), is employed to carry out genomic epidemiology. However, at very short periods of time, the core genome might not have accumulated enough variation (sufficient SNPs) to tell apart isolates. In this scenario, gene content variation in the accessory genome can be an option to conduct genomic epidemiology. Here, we used the accessory genome, as well as the core genome, to uncover the transmission dynamics of extensive and multidrug-resistant A. baumannii in a tertiary hospital for a decade. Our study shows that accessory genome variation can be a very powerful tool for conducting genomic epidemiology.
鲍曼不动杆菌已成为全球最重要的多重耐药医院病原体之一。然而,对于医院环境中并存的鲍曼不动杆菌谱系的多样性,人们了解甚少。在此,我们利用全基因组测序、流行病学数据和抗菌药敏试验,揭示了一家三级医院十年来广泛耐药和多重耐药鲍曼不动杆菌的传播动态。我们对近300个基因组的核心基因组系统发育分析表明,国际克隆2谱系多次传入该医院。分子年代分析显示,这些传入事件发生在2006年、2007年和2013年。此外,利用辅助基因组,我们发现这些谱系在医院的许多病房中广泛传播。我们的结果表明,辅助基因组变异可以成为开展基因组流行病学研究的有力工具。我们预计未来的研究将采用辅助基因组与核心基因组相结合的强大系统发育基因组学策略,在非常短的微观进化尺度上追踪细菌传播。全基因组测序用于流行病学调查(基因组流行病学)对于理解许多细菌(和非细菌)病原体的传播动态至关重要。通常,核心基因组中的变异,即单核苷酸多态性(SNP),被用于开展基因组流行病学研究。然而,在非常短的时间内,核心基因组可能没有积累足够的变异(足够的SNP)来区分分离株。在这种情况下,辅助基因组中的基因含量变异可以作为开展基因组流行病学研究的一种选择。在此,我们利用辅助基因组以及核心基因组,揭示了一家三级医院十年来广泛耐药和多重耐药鲍曼不动杆菌的传播动态。我们的研究表明,辅助基因组变异可以成为开展基因组流行病学研究的有力工具。