Conti Francesca, Catelli Arianna, Cifaldi Cristina, Leonardi Lucia, Mulè Rita, Fusconi Marco, Stefoni Vittorio, Chiriaco Maria, Rivalta Beatrice, Di Cesare Silvia, Schifino Gioacchino, Sbrega Fabiana, Di Matteo Gigliola, Ferrari Simona, Cancrini Caterina, Pession Andrea
Pediatric Unit, Istituti di Ricovero e Cura a Carattere Scientifico Azienda Ospedaliero-Universitaria di Bologna, University of Bologna, Bologna, Italy.
Specialty School of Paediatrics - Alma Mater Studiorum, University of Bologna, Bologna, Italy.
Front Pediatr. 2021 Jul 8;9:702546. doi: 10.3389/fped.2021.702546. eCollection 2021.
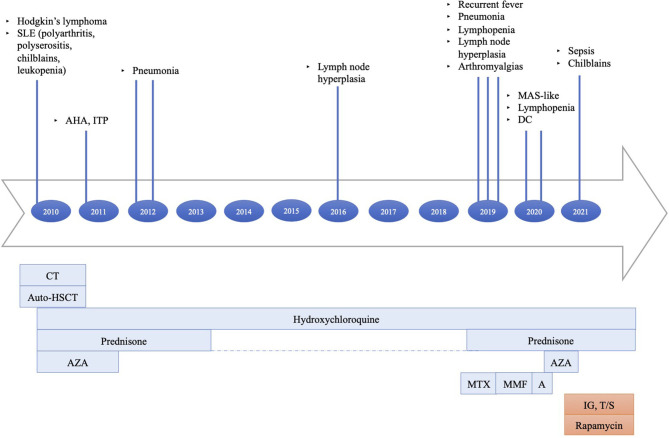
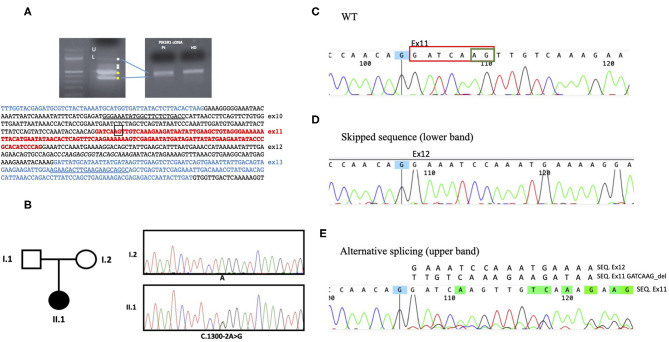
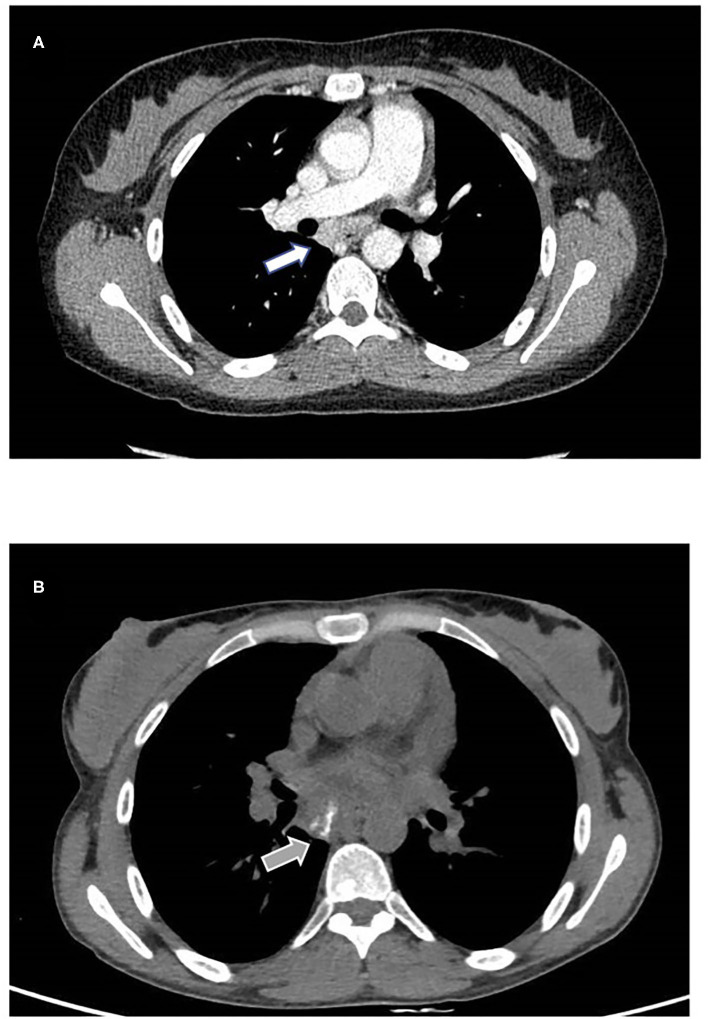
Activated phosphoinositide 3-kinase-δ syndrome 2 (APDS2) is a rare primary immune regulatory disorder caused by heterozygous gain of function mutation in the gene encoding PI3Kδ regulatory p85α subunit and resulting in PI3Kδ hyperactivation. Clinical features range from recurrent infections to manifestations of immune dysregulation like autoimmunity, inflammation, systemic lymphoproliferation, and increased risk of cancer. We describe a new dominant mutation causing APDS2 presenting with lymphoma and systemic refractory autoimmunity. A 30-year-old woman was referred to the Immunology Unit of our hospital for uncontrolled systemic lupus erythematosus, including chilblains lesions, systemic lymphoproliferation and IgA deficiency. At 19 years of age, she was diagnosed with Hodgkin's lymphoma. Subsequently, she presented systemic lupus erythematosus onset, with episodes of severe exacerbation, including autoimmune hemolytic anemia and pleuro-pericarditis. Initial clinical response to conventional treatments was reported. Immunological investigations performed during our first observation showed severe lymphopenia, IgA deficiency, elevated IgM with reduced IgG2 levels, and low vaccination antibody titers. Quantitative real-time polymerase chain reaction (PCR) assay for Cytomegalovirus and Epstein-Barr virus showed low viral loads for both viruses in serum. An increase of serum inflammatory markers highlighted persistent systemic hyperinflammation. The next-generation sequencing (NGS)-based gene panel tests for primary immunodeficiency showed a heterozygous A>G substitution in the splice acceptor site at c.1300-2 position of , leading to exon-skipping. This case emphasizes the importance of suspecting primary immune regulatory disorders in young adults, predominantly showing a severe, aggressive, and refractory to treatment immune dysregulation phenotype, even in the absence of major infectious diseases at the onset. Different treatments can be promptly started, and a delayed diagnosis can highly impact the outcome. Targeted therapy against PI3Kδ pathway defect effectively improves drug-resistant autoimmunity, lymphoproliferation, and risk of progression to malignancy; eligible patients could benefit from its use even as a bridge therapy to transplantation, currently the only definitive curative treatment. Therefore, identifying genetic mutation and prompt targeted treatment are essential to control disease manifestations, prevent long-term sequelae, and enable curative HSCT in APDS2 patients.
活化磷脂酰肌醇3-激酶δ综合征2(APDS2)是一种罕见的原发性免疫调节障碍,由编码PI3Kδ调节性p85α亚基的基因杂合性功能获得性突变引起,导致PI3Kδ过度活化。临床特征从反复感染到免疫失调表现,如自身免疫、炎症、全身性淋巴细胞增殖和癌症风险增加。我们描述了一种导致APDS2的新显性突变,表现为淋巴瘤和全身性难治性自身免疫。一名30岁女性因系统性红斑狼疮控制不佳被转诊至我院免疫科,包括冻疮样皮损、全身性淋巴细胞增殖和IgA缺乏。19岁时,她被诊断为霍奇金淋巴瘤。随后,她出现系统性红斑狼疮发作,伴有严重加重期,包括自身免疫性溶血性贫血和胸膜心包炎。报告了对传统治疗的初始临床反应。我们首次观察期间进行的免疫学检查显示严重淋巴细胞减少、IgA缺乏、IgM升高伴IgG2水平降低以及疫苗接种抗体滴度低。巨细胞病毒和EB病毒的定量实时聚合酶链反应(PCR)检测显示血清中两种病毒的病毒载量均低。血清炎症标志物升高突出了持续的全身性高炎症。基于下一代测序(NGS)的原发性免疫缺陷基因panel检测显示,在 基因第c.1300-2位的剪接受体位点存在杂合性A>G替换,导致外显子跳跃。该病例强调了在年轻人中怀疑原发性免疫调节障碍的重要性,这些年轻人主要表现出严重、侵袭性且对治疗难治的免疫失调表型,即使在发病时没有主要传染病。可以迅速开始不同的治疗,而延迟诊断会对结果产生重大影响。针对PI3Kδ通路缺陷的靶向治疗可有效改善耐药性自身免疫、淋巴细胞增殖以及进展为恶性肿瘤的风险;符合条件的患者即使作为移植的桥接治疗也可从中受益,目前移植是唯一明确的治愈性治疗方法。因此,识别基因突变并及时进行靶向治疗对于控制疾病表现、预防长期后遗症以及使APDS2患者能够进行治愈性造血干细胞移植至关重要。