McLay Todd G B, Birch Joanne L, Gunn Bee F, Ning Weixuan, Tate Jennifer A, Nauheimer Lars, Joyce Elizabeth M, Simpson Lalita, Schmidt-Lebuhn Alexander N, Baker William J, Forest Félix, Jackson Chris J
National Herbarium of Victoria Royal Botanic Gardens Victoria Melbourne Australia.
School of Biosciences University of Melbourne Melbourne Australia.
Appl Plant Sci. 2021 Jun 14;9(7). doi: 10.1002/aps3.11420. eCollection 2021 Jul.
Universal target enrichment kits maximize utility across wide evolutionary breadth while minimizing the number of baits required to create a cost-efficient kit. The Angiosperms353 kit has been successfully used to capture loci throughout the angiosperms, but the default target reference file includes sequence information from only 6-18 taxa per locus. Consequently, reads sequenced from on-target DNA molecules may fail to map to references, resulting in fewer on-target reads for assembly, and reducing locus recovery.
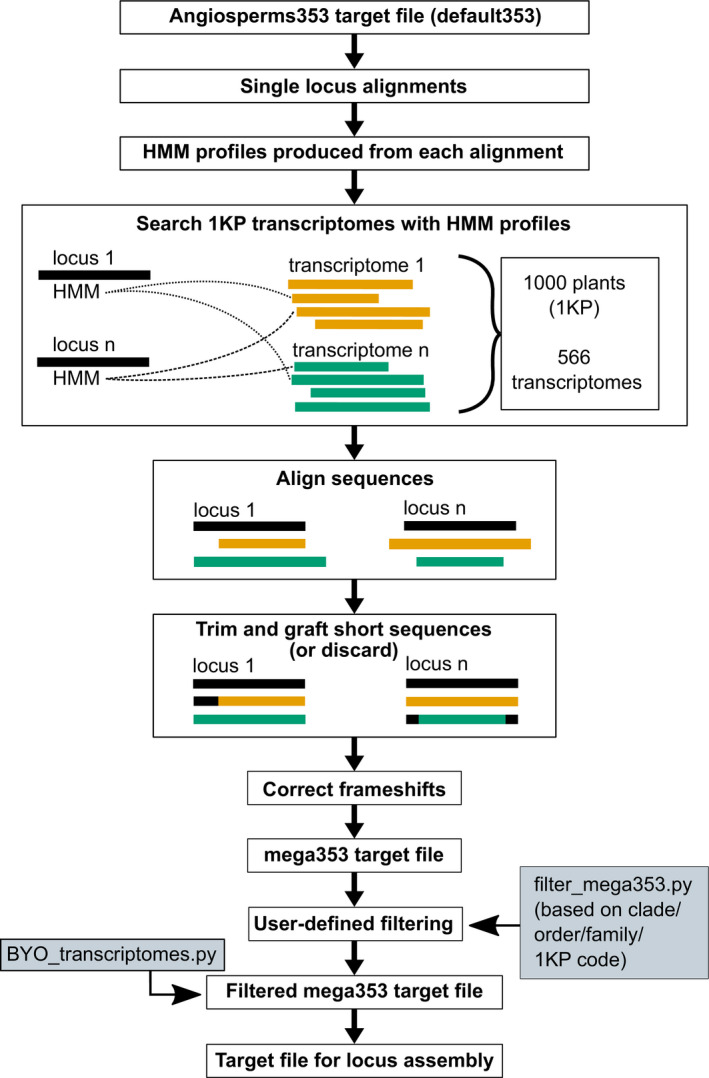
We expanded the Angiosperms353 target file, incorporating sequences from 566 transcriptomes to produce a 'mega353' target file, with each locus represented by 17-373 taxa. This mega353 file is a drop-in replacement for the original Angiosperms353 file in HybPiper analyses. We provide tools to subsample the file based on user-selected taxon groups, and to incorporate other transcriptome or protein-coding gene data sets.
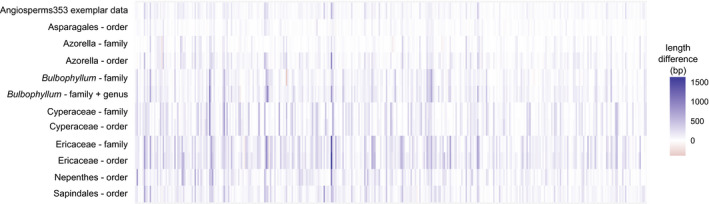
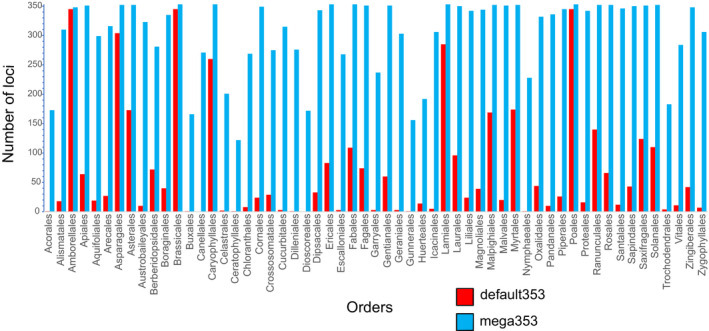
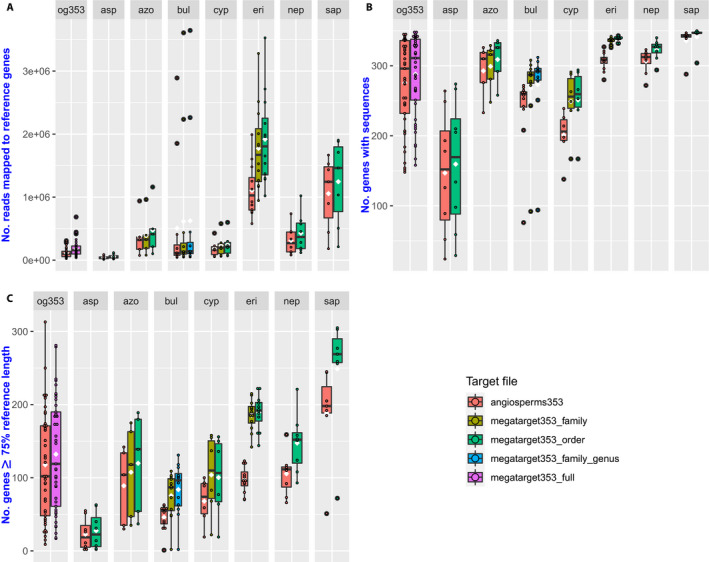
Compared to the default Angiosperms353 file, the mega353 file increased the percentage of on-target reads by an average of 32%, increased locus recovery at 75% length by 49%, and increased the total length of the concatenated loci by 29%.
Increasing the phylogenetic density of the target reference file results in improved recovery of target capture loci. The mega353 file and associated scripts are available at: https://github.com/chrisjackson-pellicle/NewTargets.
通用目标富集试剂盒在广泛的进化广度上最大限度地提高了实用性,同时将创建具有成本效益的试剂盒所需的诱饵数量降至最低。被子植物353试剂盒已成功用于捕获整个被子植物的基因座,但默认的目标参考文件每个基因座仅包含6 - 18个分类群的序列信息。因此,从靶向DNA分子测序得到的 reads 可能无法映射到参考序列,导致用于组装的靶向 reads 减少,进而降低基因座回收率。
我们扩展了被子植物353目标文件,纳入了566个转录组的序列,以生成一个“mega353”目标文件,每个基因座由17 - 373个分类群表示。这个 mega353 文件可直接替代HybPiper分析中原来的被子植物353文件。我们提供了工具,可根据用户选择的分类群进行文件抽样,并纳入其他转录组或蛋白质编码基因数据集。
与默认的被子植物353文件相比,mega353文件使靶向 reads 的百分比平均提高了32%,75%长度的基因座回收率提高了49%,串联基因座的总长度增加了29%。
增加目标参考文件的系统发育密度可提高目标捕获基因座的回收率。mega353文件及相关脚本可在以下网址获取:https://github.com/chrisjackson-pellicle/NewTargets。