Division of Ecology and Evolution, Research School of Biology, The Australian National University, Canberra, Australian Capital Territory, Australia.
The University of Queensland, School of Biological Sciences, Brisbane, Queensland, Australia.
PLoS One. 2019 Aug 1;14(8):e0218995. doi: 10.1371/journal.pone.0218995. eCollection 2019.
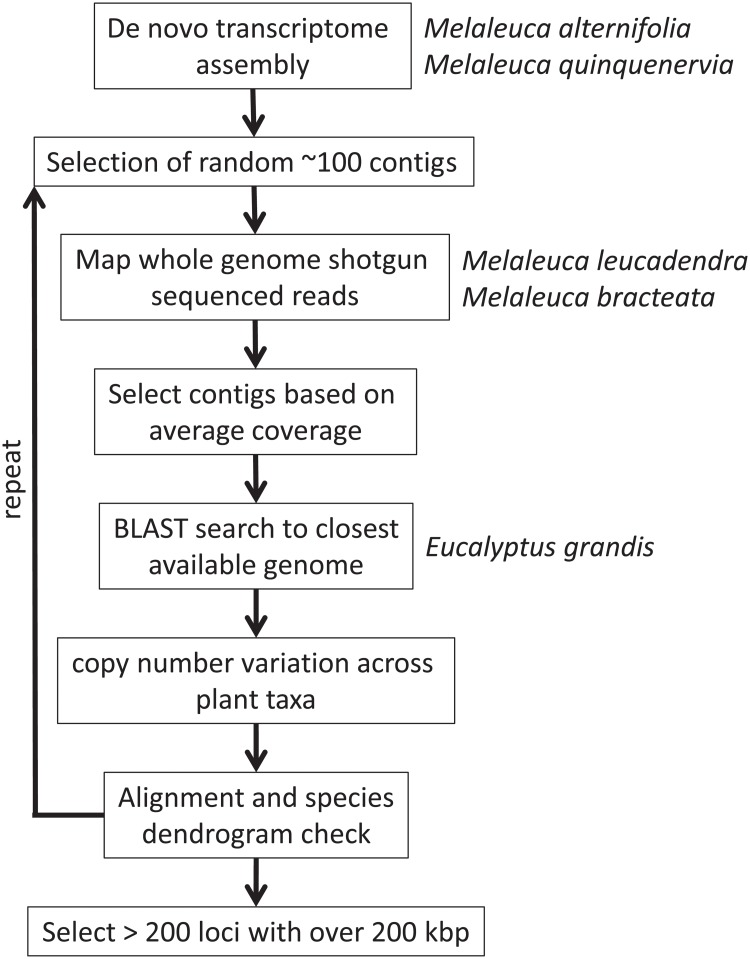
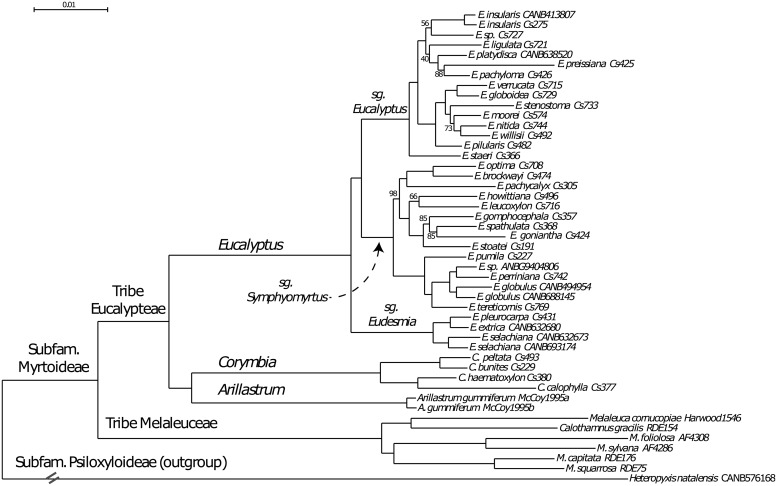
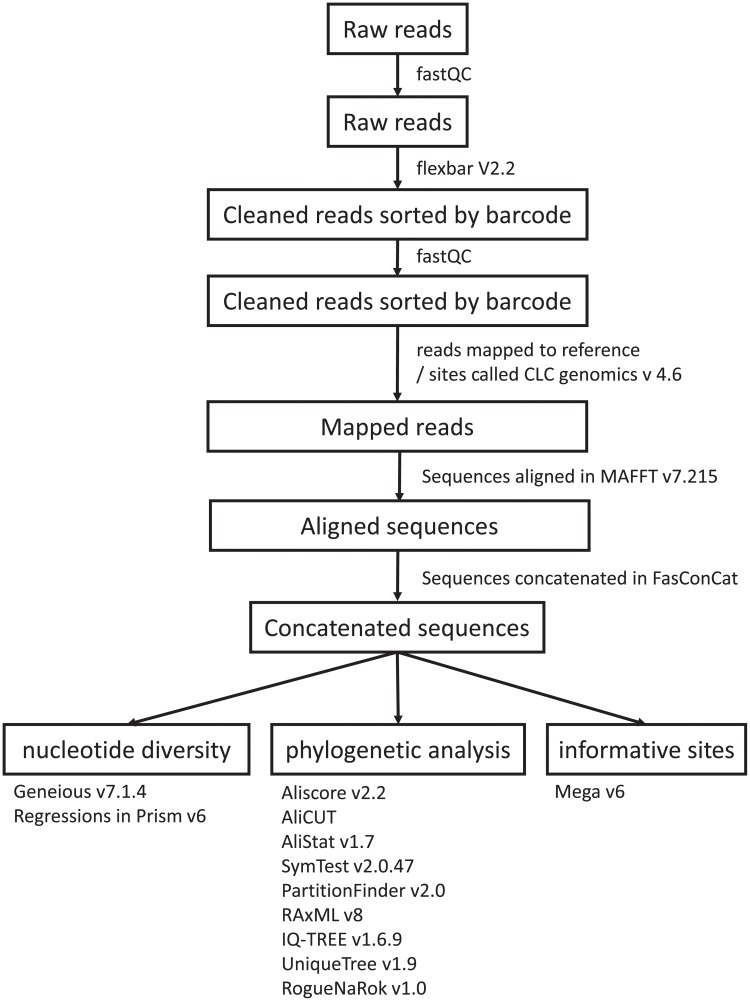
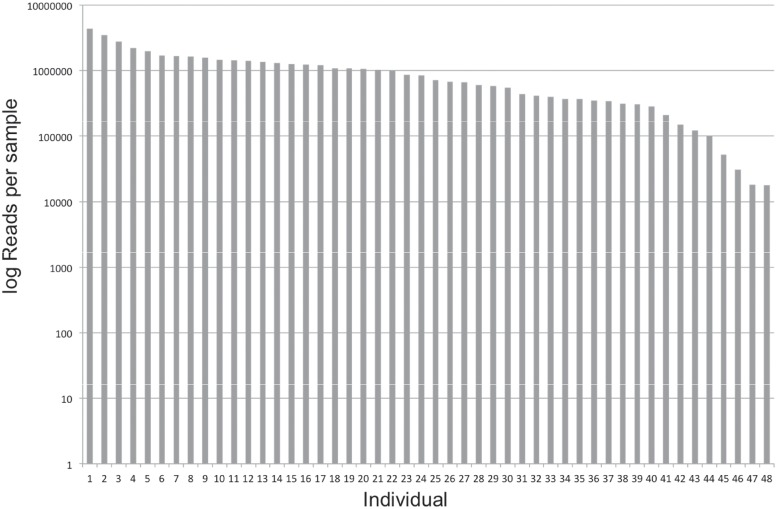
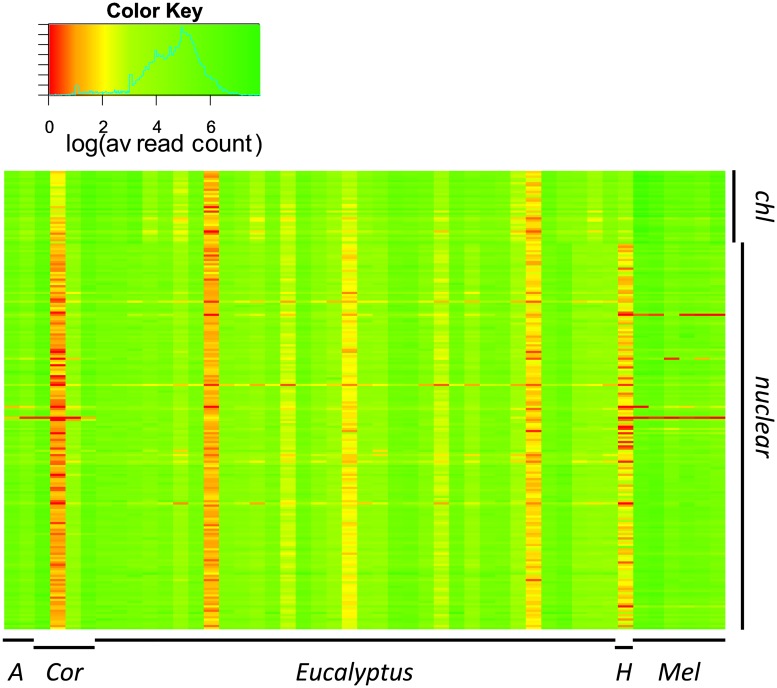
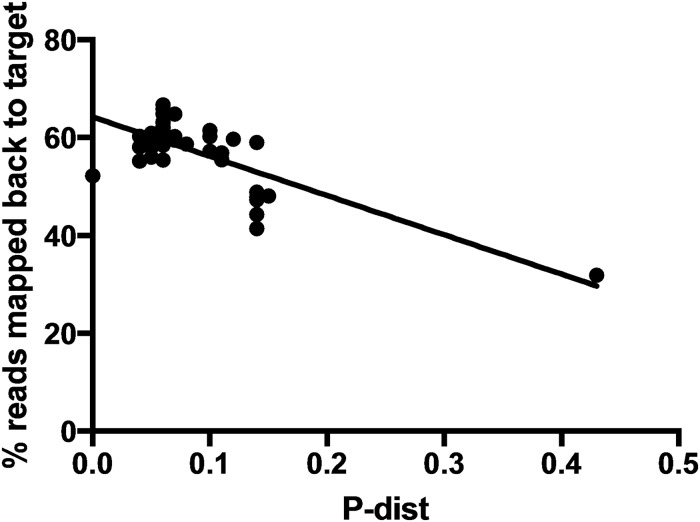
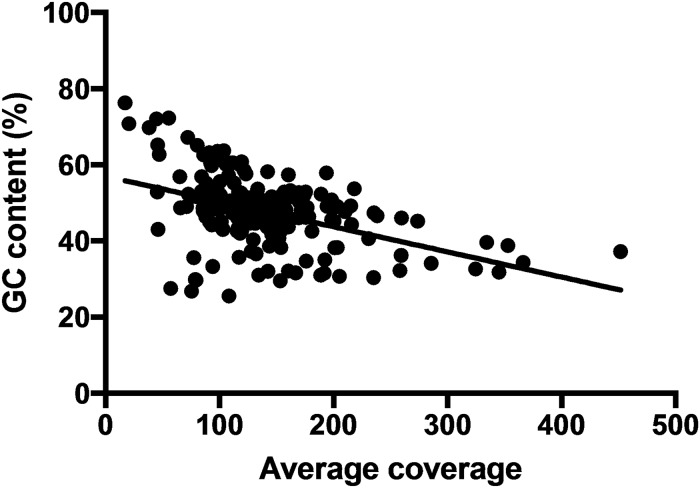
Resolving the phylogenetic relationships of closely related species using a small set of loci is challenging as sufficient information may not be captured from a limited sample of the genome. Relying on few loci can also be problematic when conflict between gene-trees arises from incomplete lineage sorting and/or ongoing hybridization, problems especially likely in recently diverged lineages. Here, we developed a method using limited genomic resources that allows identification of many low copy candidate loci from across the nuclear and chloroplast genomes, design probes for target capture and sequence the captured loci. To validate our method we present data from Eucalyptus and Melaleuca, two large and phylogenetically problematic genera within the Myrtaceae family. With one annotated genome, one transcriptome and two whole-genome shotgun sequences of one Eucalyptus and four Melaleuca species, respectively, we identified 212 loci representing 263 kbp for targeted sequence capture and sequencing. Of these, 209 were successfully tested from 47 samples across five related genera of Myrtaceae. The average percentage of reads mapped back to the reference was 57.6% with coverage of more than 20 reads per position across 83.5% of the data. The methods developed here should be applicable across a large range of taxa across all kingdoms. The core methods are very flexible, providing a platform for various genomic resource availabilities and are useful from shallow to deep phylogenies.
使用少量基因座来解决近缘物种的系统发育关系具有挑战性,因为从基因组的有限样本中可能无法获取足够的信息。当基因树之间的冲突来自不完全谱系分选和/或正在进行的杂交时,依赖少数基因座也会出现问题,这种问题在最近分化的谱系中尤为明显。在这里,我们开发了一种利用有限基因组资源的方法,该方法允许从核和叶绿体基因组中识别许多低拷贝的候选基因座,设计目标捕获的探针并对捕获的基因座进行测序。为了验证我们的方法,我们展示了来自桃金娘科的桉树和千层树两个大的和系统发育上有问题的属的数据。我们使用一个注释基因组、一个转录组和一个桉树和四个千层树物种的全基因组鸟枪法序列,分别鉴定了 212 个代表 263 kbp 的基因座,用于靶向序列捕获和测序。其中,在桃金娘科的五个相关属的 47 个样本中成功测试了 209 个。在 83.5%的数据中,每个位置的覆盖度超过 20 个读数,平均有 57.6%的读数映射回参考基因组。这里开发的方法应该适用于所有生物领域的大量分类群。核心方法非常灵活,为各种基因组资源的可用性提供了一个平台,并且从浅层到深层系统发育都非常有用。