Department of Pharmacology and Toxicology, Faculty of Pharmacy, Comenius University in Bratislava, 81499 Bratislava, Slovakia.
Faculty of Health Sciences, Bristol Heart Institute, The Bristol Medical School, University of Bristol, Bristol BS8 1TH, UK.
Int J Mol Sci. 2021 Jul 26;22(15):7983. doi: 10.3390/ijms22157983.
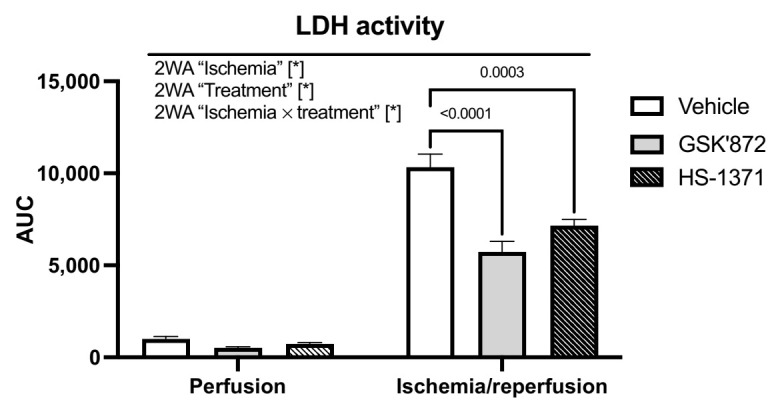
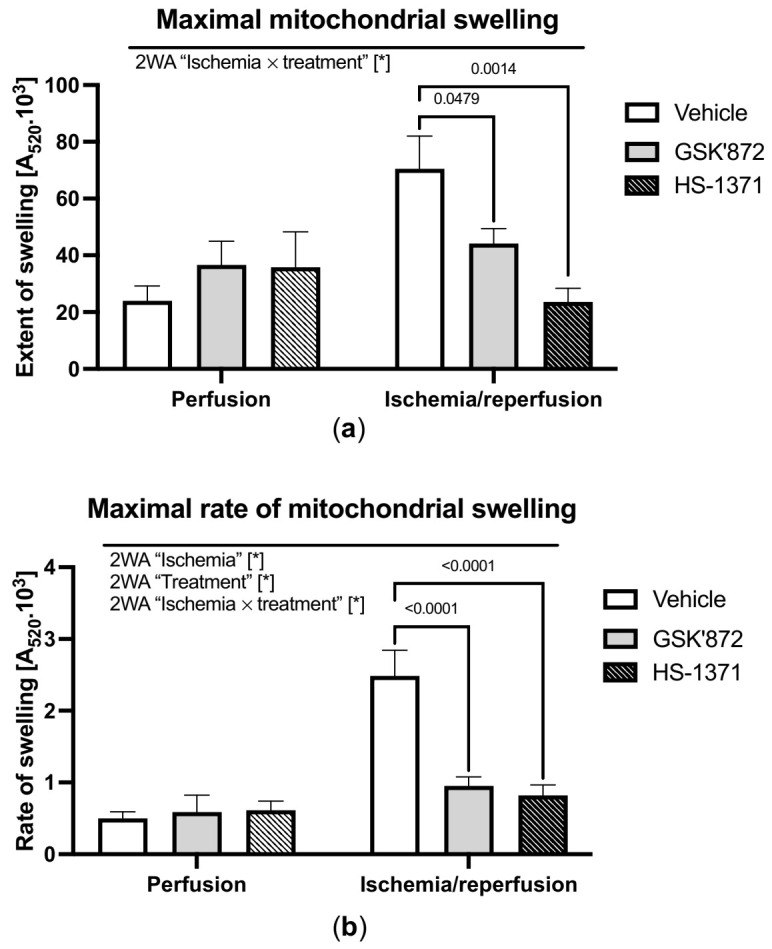
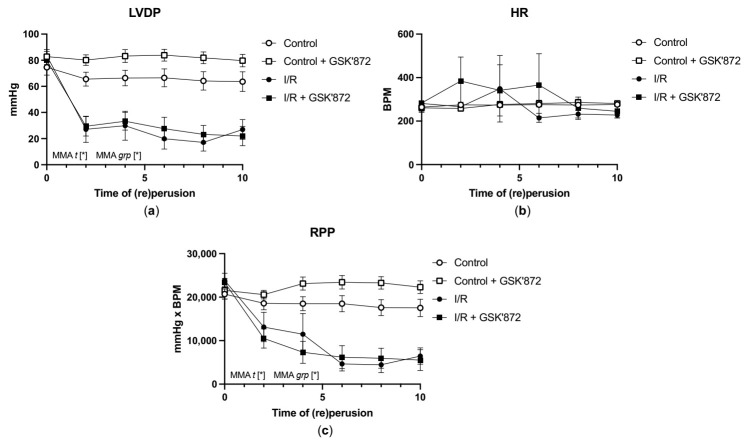
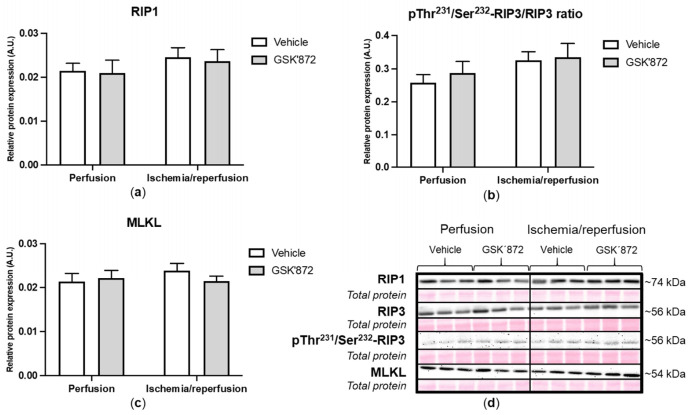
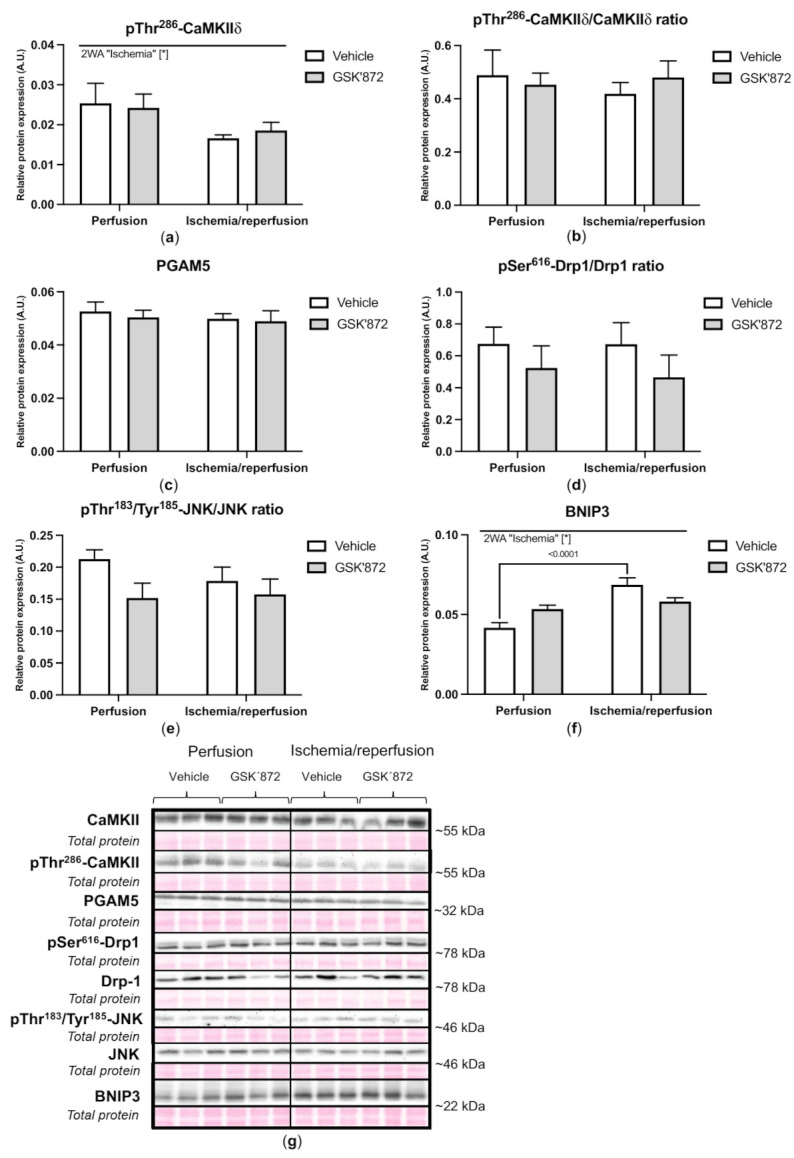
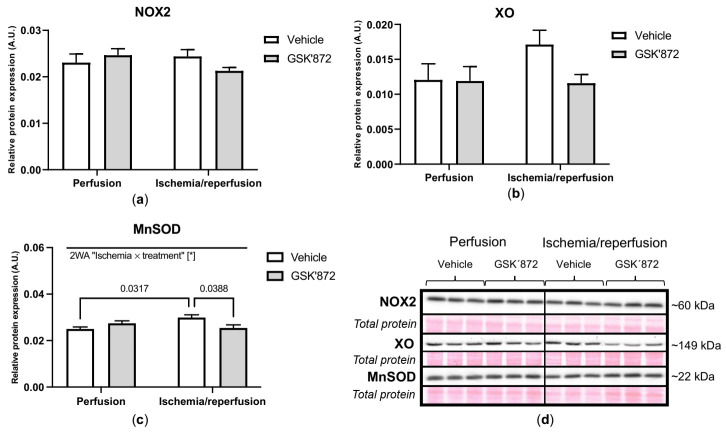
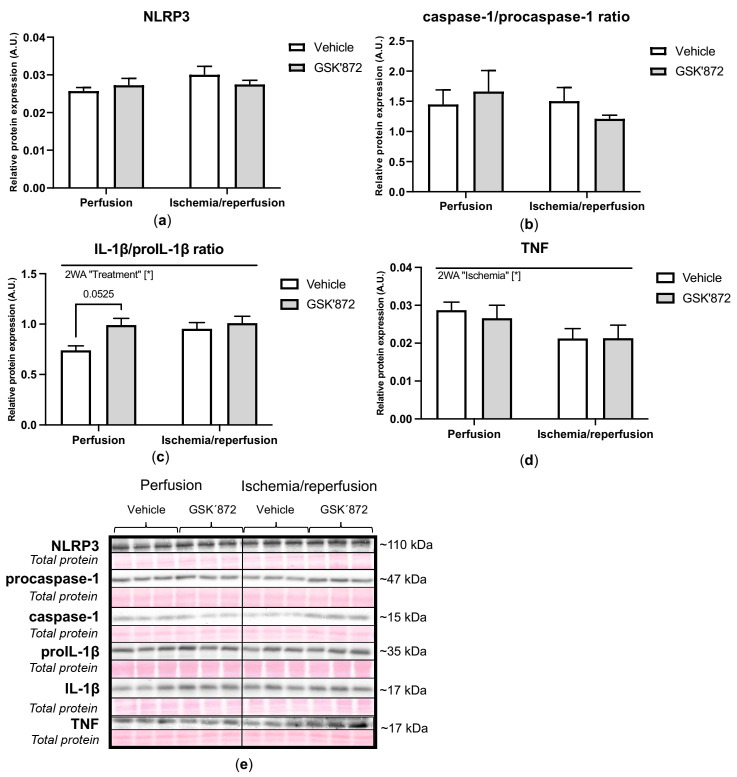
Receptor-interacting protein kinase 3 (RIP3) is a convergence point of multiple signalling pathways, including necroptosis, inflammation and oxidative stress; however, it is completely unknown whether it underlies acute myocardial ischemia/reperfusion (I/R) injury. Langendorff-perfused rat hearts subjected to 30 min ischemia followed by 10 min reperfusion exhibited compromised cardiac function which was not abrogated by pharmacological intervention of RIP3 inhibition. An immunoblotting analysis revealed that the detrimental effects of I/R were unlikely mediated by necroptotic cell death, since neither the canonical RIP3-MLKL pathway (mixed lineage kinase-like pseudokinase) nor the proposed non-canonical molecular axes involving CaMKIIδ-mPTP (calcium/calmodulin-dependent protein kinase IIδ-mitochondrial permeability transition pore), PGAM5-Drp1 (phosphoglycerate mutase 5-dynamin-related protein 1) and JNK-BNIP3 (c-Jun N-terminal kinase-BCL2-interacting protein 3) were activated. Similarly, we found no evidence of the involvement of NLRP3 inflammasome signalling (NOD-, LRR- and pyrin domain-containing protein 3) in such injury. RIP3 inhibition prevented the plasma membrane rupture and delayed mPTP opening which was associated with the modulation of xanthin oxidase (XO) and manganese superoxide dismutase (MnSOD). Taken together, this is the first study indicating that RIP3 regulates early reperfusion injury via oxidative stress- and mitochondrial activity-related effects, rather than cell loss due to necroptosis.
受体相互作用蛋白激酶 3(RIP3)是多种信号通路的交汇点,包括坏死性凋亡、炎症和氧化应激;然而,它是否是急性心肌缺血/再灌注(I/R)损伤的基础完全未知。Langendorff 灌注大鼠心脏缺血 30 分钟后再灌注 10 分钟,表现出心脏功能受损,而 RIP3 抑制的药物干预并不能消除这种损伤。免疫印迹分析表明,I/R 的有害作用不太可能是通过坏死性细胞死亡介导的,因为既没有经典的 RIP3-MLKL 途径(混合谱系激酶样伪激酶),也没有涉及 CaMKIIδ-mPTP(钙/钙调蛋白依赖性蛋白激酶 IIδ-线粒体通透性转换孔)、PGAM5-Drp1(磷酸甘油酸变位酶 5-动力蛋白相关蛋白 1)和 JNK-BNIP3(c-Jun N 末端激酶-BCL2 相互作用蛋白 3)的拟议非经典分子轴被激活。同样,我们没有发现 NLRP3 炎性体信号(含 NOD、LRR 和吡喃结构域蛋白 3)参与这种损伤的证据。RIP3 抑制阻止了质膜破裂和 mPTP 开放的延迟,这与黄嘌呤氧化酶(XO)和锰超氧化物歧化酶(MnSOD)的调节有关。总之,这是第一项表明 RIP3 通过与氧化应激和线粒体活性相关的作用调节早期再灌注损伤,而不是由于坏死性凋亡导致细胞死亡的研究。