iThree Institute, University of Technology Sydney, Sydney, New South Wales, Australia.
NSW Department of Primary Industries, Elizabeth Macarthur Agricultural Institute, Menangle, New South Wales, Australia.
Microb Genom. 2021 Aug;7(8). doi: 10.1099/mgen.0.000501.
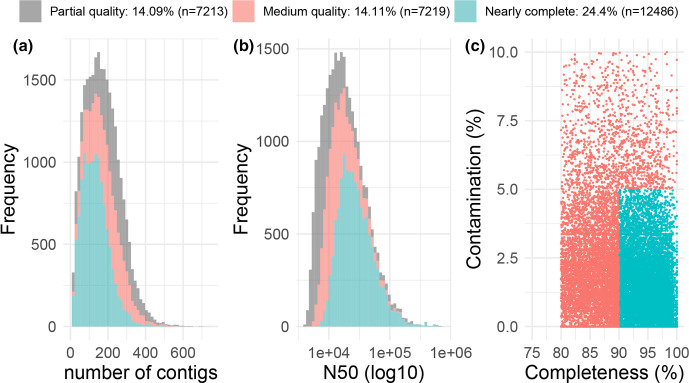
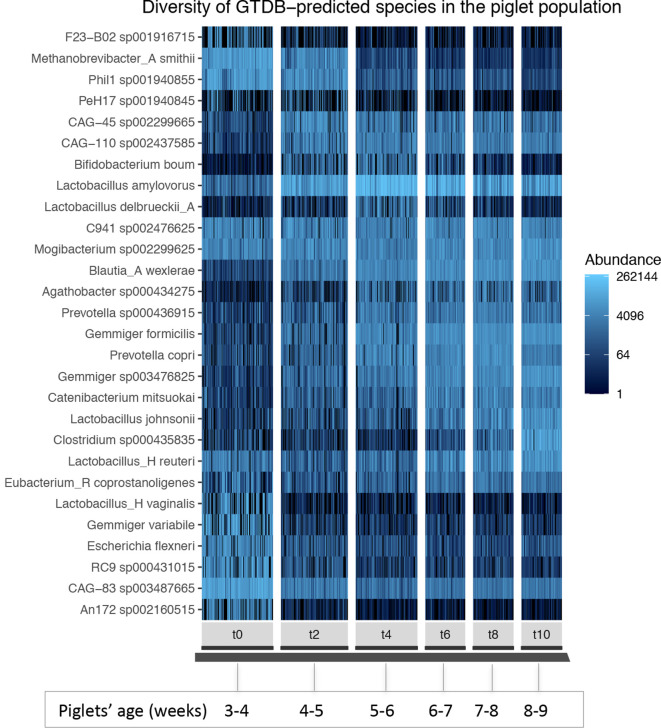
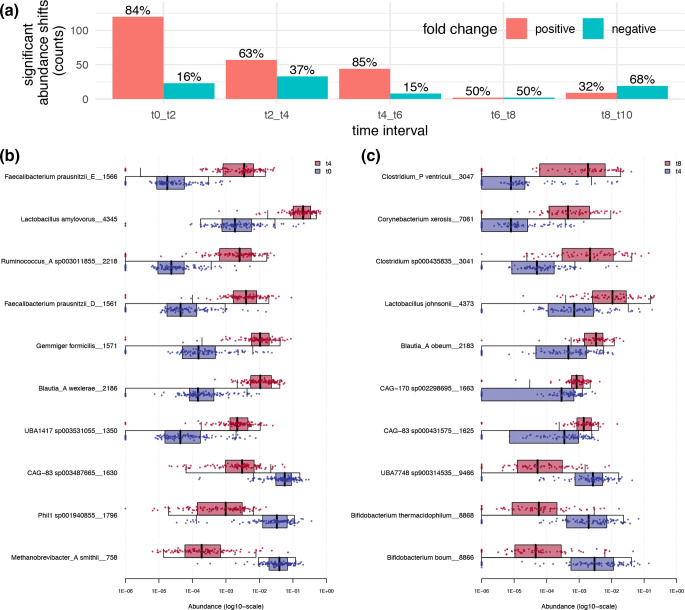
Using a previously described metagenomics dataset of 27 billion reads, we reconstructed over 50 000 metagenome-assembled genomes (MAGs) of organisms resident in the porcine gut, 46.5 % of which were classified as >70 % complete with a <10 % contamination rate, and 24.4 % were nearly complete genomes. Here, we describe the generation and analysis of those MAGs using time-series samples. The gut microbial communities of piglets appear to follow a highly structured developmental programme in the weeks following weaning, and this development is robust to treatments including an intramuscular antibiotic treatment and two probiotic treatments. The high resolution we obtained allowed us to identify specific taxonomic 'signatures' that characterize the gut microbial development immediately after weaning. Additionally, we characterized the carbohydrate repertoire of the organisms resident in the porcine gut. We tracked the abundance shifts of 294 carbohydrate active enzymes, and identified the species and higher-level taxonomic groups carrying each of these enzymes in their MAGs. This knowledge can contribute to the design of probiotics and prebiotic interventions as a means to modify the piglet gut microbiome.
使用先前描述的 270 亿个读数的宏基因组数据集,我们重建了超过 50000 个居住在猪肠道中的生物体的宏基因组组装基因组(MAG),其中 46.5%的分类为> 70%完整,污染率< 10%,并且 24.4%的是近乎完整的基因组。在这里,我们使用时间序列样本描述了这些 MAG 的生成和分析。仔猪肠道微生物群落似乎在断奶后的几周内遵循一个高度结构化的发育计划,这种发育对包括肌肉内抗生素治疗和两种益生菌治疗在内的处理具有很强的稳健性。我们获得的高分辨率使我们能够识别出在断奶后立即表征肠道微生物发育的特定分类“特征”。此外,我们还描述了猪肠道中居住的生物体的碳水化合物组成。我们跟踪了 294 种碳水化合物活性酶的丰度变化,并确定了在其 MAG 中携带每种这些酶的物种和更高分类群。这些知识可以为益生菌和益生元干预措施的设计做出贡献,作为改变仔猪肠道微生物组的一种手段。