European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton, Cambridge, United Kingdom.
Mol Genet Genomic Med. 2021 Dec;9(12):e1786. doi: 10.1002/mgg3.1786. Epub 2021 Aug 26.
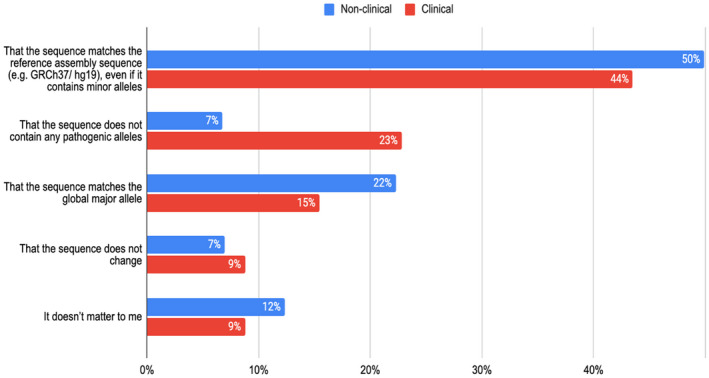
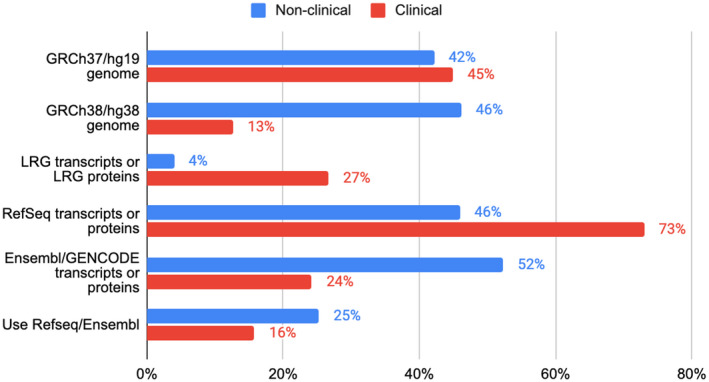
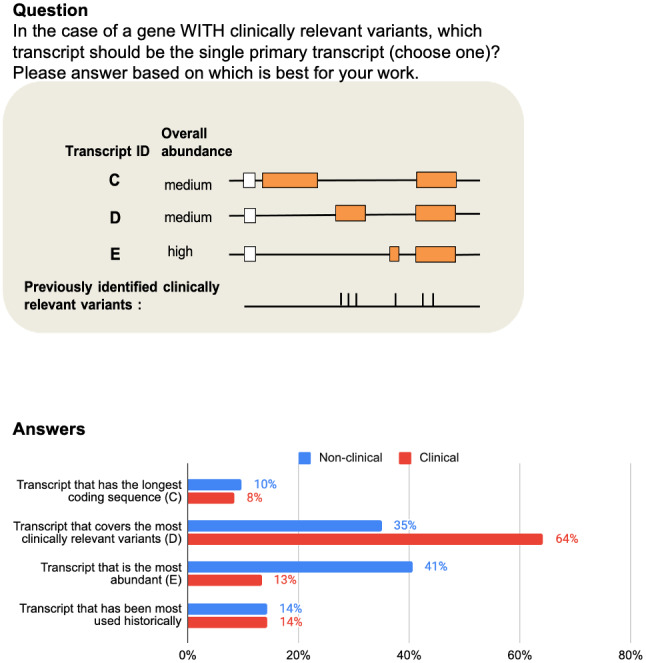
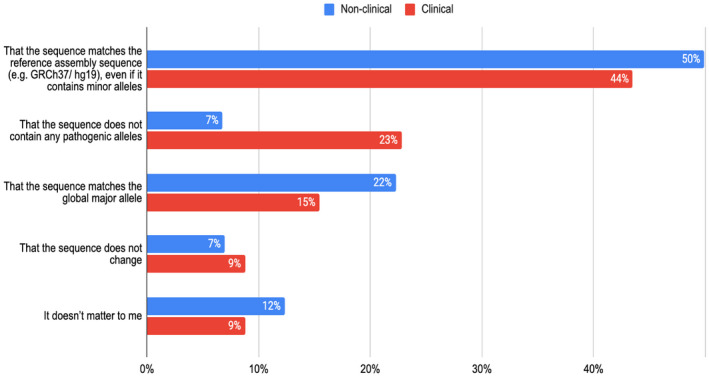
Variant interpretation is dependent on transcript annotation and remains time consuming and challenging. There are major obstacles for historical data reuse and for interpretation of new variants. First, both RefSeq and Ensembl/GENCODE produce transcript sets in common use, but there is currently no easy way to translate between the two. Second, the resources often used for variant interpretation (e.g. ClinVar, gnomAD, UniProt) do not use the same transcript set, nor default transcript or protein sequence.
Ensembl ran a survey in 2018 to sample attitudes to choosing one default transcript per locus, and to gather data on reference sequences used by the scientific community. This was publicised on the Ensembl and UCSC genome browsers, by email and on social media.
The survey had 788 responses from 32 different countries, the results of which we report here.
We present our roadmap to create an effective default set of transcripts for resources, and for reporting interpretation of clinical variants.
变体解释依赖于转录本注释,仍然耗时且具有挑战性。历史数据的重复使用和新变体的解释存在重大障碍。首先,RefSeq 和 Ensembl/GENCODE 都生成常用的转录本集,但目前没有简便的方法在两者之间进行转换。其次,变体解释常用的资源(例如 ClinVar、gnomAD、UniProt)不使用相同的转录本集,也没有默认的转录本或蛋白质序列。
Ensembl 在 2018 年进行了一项调查,以抽样选择每个基因座的一个默认转录本的态度,并收集有关科学界使用的参考序列的数据。该调查在 Ensembl 和 UCSC 基因组浏览器、电子邮件和社交媒体上进行了宣传。
该调查收到了来自 32 个不同国家的 788 份回复,我们在此报告这些回复的结果。
我们提出了创建资源和报告临床变体解释的有效默认转录本集的路线图。