Zhou Wangxiao, Jin Ye, Zhou Yanzi, Wang Yuan, Xiong Luying, Luo Qixia, Xiao Yonghong
State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
mSystems. 2021 Oct 26;6(5):e0098621. doi: 10.1128/mSystems.00986-21. Epub 2021 Sep 7.
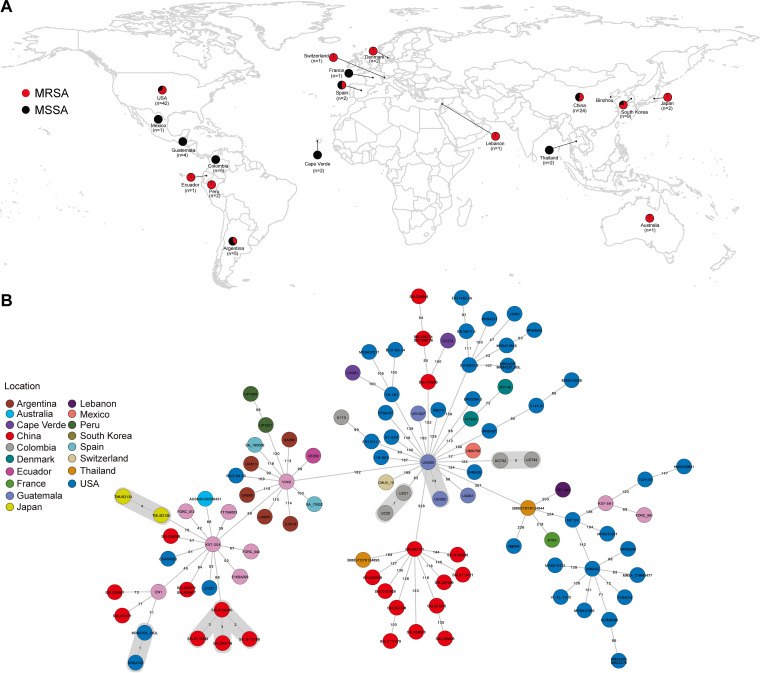
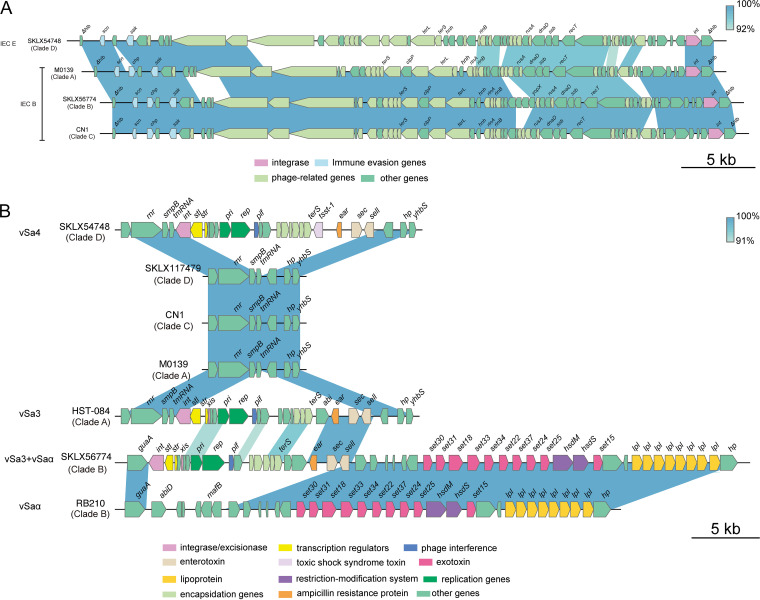
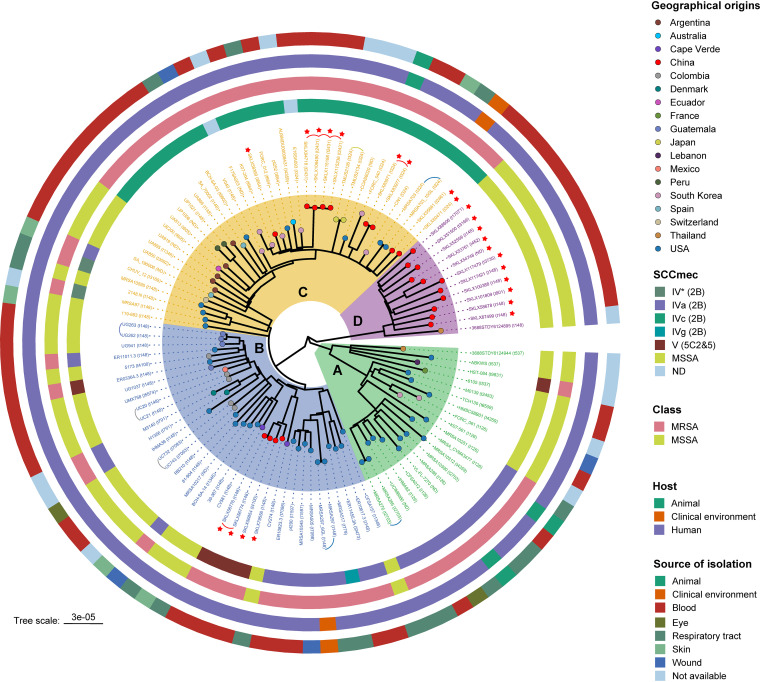
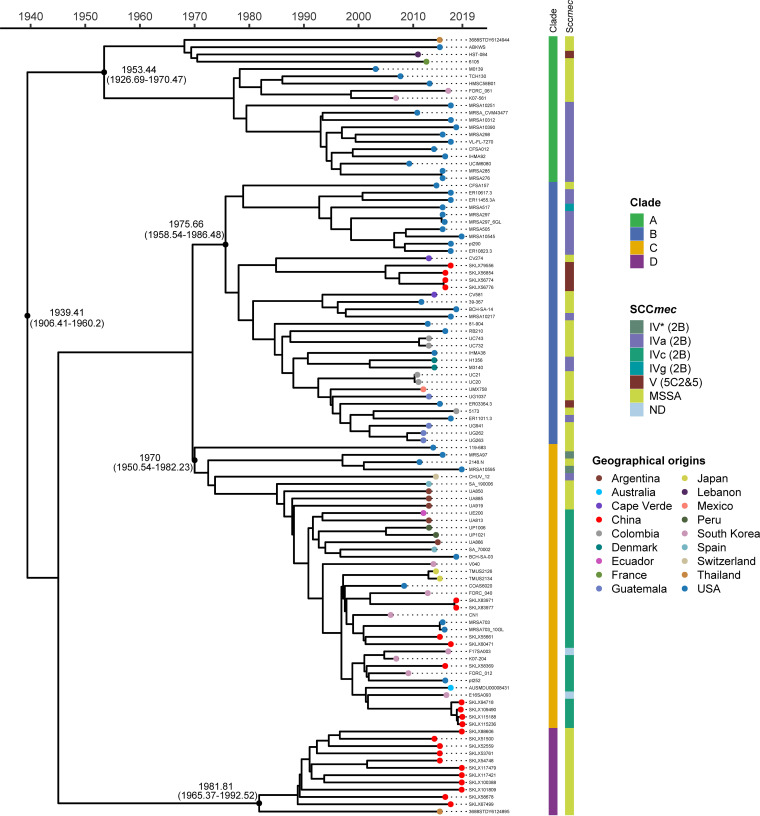
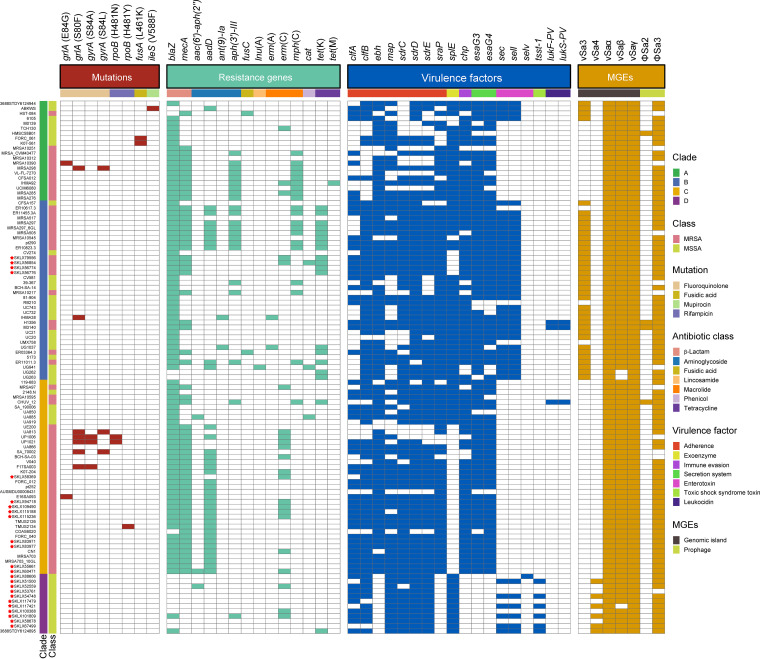
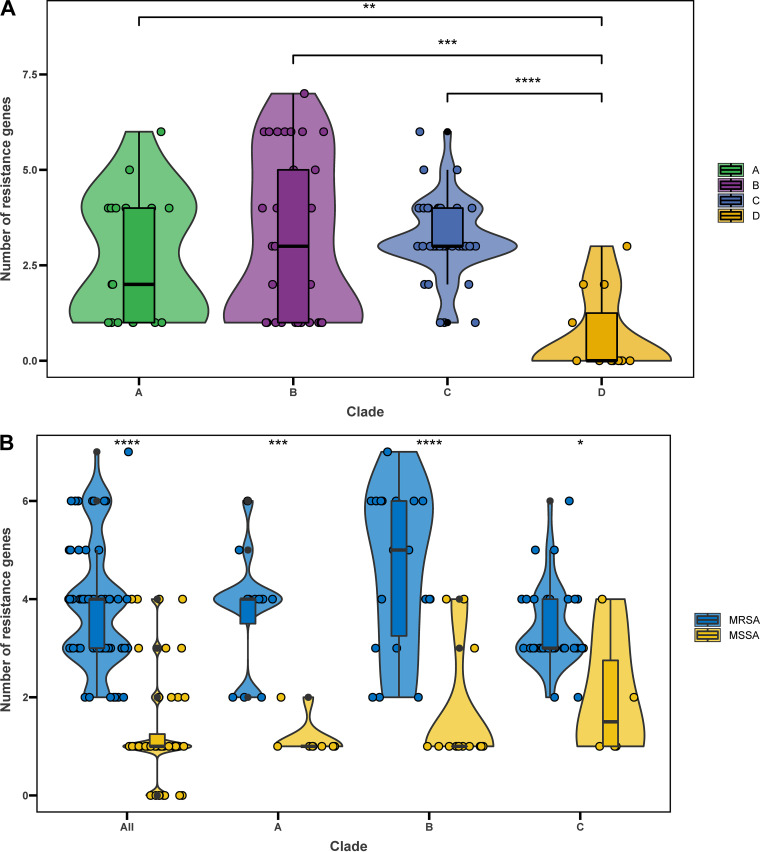
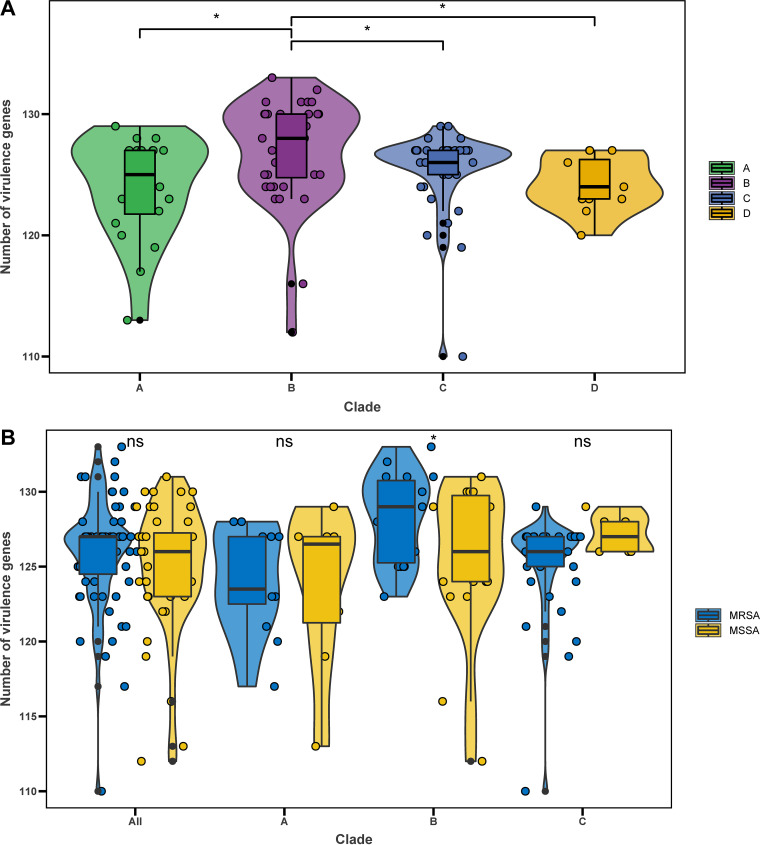
Staphylococcus aureus sequence type (ST) 72, the predominant community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA) lineage in South Korea, has emerged as a major cause of bloodstream infection in hospital settings. However, relatively little information is available regarding the genomic characteristics and dissemination of ST72. Here, we characterized the whole-genome sequence of 24 ST72 isolates from China, along with 83 ST72 genomes from global sources. Of these 107 ST72 isolates, 63 were MRSA and 44 were methicillin-susceptible S. aureus (MSSA). Phylogenetic analysis revealed four distinct clades (A, B, C, and D), of which clade D contained only MSSA isolates. By characterizing the evolutionary dynamics of the ST72 lineage, we found that the MRSA from China might not have developed from the MSSA in China. Furthermore, we observed both international transmission of ST72 isolates and interregional transmission within China. The distributions of the SCC and types of isolates differed among clades. Additionally, analyses revealed that the distributions of resistance genes, virulence genes, and mobile genetic elements (MGEs) also differed among isolates of the four clades. This was especially true for clade D isolates, which had the lowest level of antimicrobial resistance and had obtained specific virulence genes such as by acquisition of specific MGEs. Notably, ST72 MRSA isolates were more antibiotic resistant than ST72 MSSA isolates, but comparably virulent. Our findings provide insight into the potential transmission and genotypic features of ST72 clones across the globe. Understanding the evolution and dissemination of community-genotype ST72 Staphylococcus aureus isolates is important, as isolates of this lineage have rapidly spread into hospital settings and caused serious health issues. In this study, we first carried out genome-wide analysis of 107 global ST72 isolates to characterize the evolution and genetic diversity of the ST72 lineage. We found that the MSSA lineage in China might have evolved independently from the MRSA isolates from China, and that ST72 isolates have the potential to undergo both international transmission and interregional transmission within China. The diversity of isolates correlated with distinct acquisitions of SCC elements, antibiotic resistance genes, virulence genes, and mobile genetic elements. The comprehensive information on the ST72 lineage emerging from this study will enable improved therapeutic approaches and rapid molecular diagnosis.
金黄色葡萄球菌序列类型(ST)72是韩国社区获得性耐甲氧西林金黄色葡萄球菌(CA-MRSA)的主要谱系,已成为医院环境中血流感染的主要原因。然而,关于ST72的基因组特征和传播的信息相对较少。在此,我们对来自中国的24株ST72分离株以及来自全球的83个ST72基因组进行了全基因组测序分析。在这107株ST72分离株中,63株为耐甲氧西林金黄色葡萄球菌(MRSA),44株为甲氧西林敏感金黄色葡萄球菌(MSSA)。系统发育分析揭示了四个不同的分支(A、B、C和D),其中分支D仅包含MSSA分离株。通过分析ST72谱系的进化动态,我们发现中国的MRSA可能并非由中国的MSSA进化而来。此外,我们观察到ST72分离株存在国际传播以及在中国境内的区域间传播。葡萄球菌盒式染色体(SCC)的分布和分离株类型在各分支之间存在差异。此外,分析表明,四个分支的分离株中耐药基因、毒力基因和移动遗传元件(MGE)的分布也有所不同。对于分支D分离株尤其如此,其抗菌耐药水平最低,并通过获取特定的MGE获得了如某些特定的毒力基因。值得注意的是,ST72 MRSA分离株比ST72 MSSA分离株具有更高的抗生素耐药性,但毒力相当。我们的研究结果为全球范围内ST72克隆的潜在传播和基因型特征提供了见解。了解社区基因型ST72金黄色葡萄球菌分离株的进化和传播很重要,因为该谱系的分离株已迅速传播到医院环境并引发严重的健康问题。在本研究中,我们首先对107株全球ST72分离株进行了全基因组分析,以描述ST72谱系的进化和遗传多样性。我们发现中国的MSSA谱系可能独立于中国的MRSA分离株进化而来,并且ST72分离株有在中国境内进行国际传播和区域间传播的潜力。分离株的多样性与SCC元件、抗生素耐药基因、毒力基因和移动遗传元件的不同获得情况相关。本研究中关于ST72谱系的全面信息将有助于改进治疗方法和快速分子诊断。