Department of Neuroscience, College of Medicine, University of Florida, Gainesville, Florida, 32610, USA.
Center for Translational Research in Neurodegenerative Disease, College of Medicine, University of Florida, Gainesville, Florida, 32610, USA.
Mol Neurodegener. 2021 Sep 9;16(1):63. doi: 10.1186/s13024-021-00486-9.
The misfolding of host-encoded proteins into pathological prion conformations is a defining characteristic of many neurodegenerative disorders, including Alzheimer's disease, Parkinson's disease, and Lewy body dementia. A current area of intense study is the way in which the pathological deposition of these proteins might influence each other, as various combinations of co-pathology between prion-capable proteins are associated with exacerbation of disease. A spectrum of pathological, genetic and biochemical evidence provides credence to the notion that amyloid β (Aβ) accumulation can induce and promote α-synuclein pathology, driving neurodegeneration.
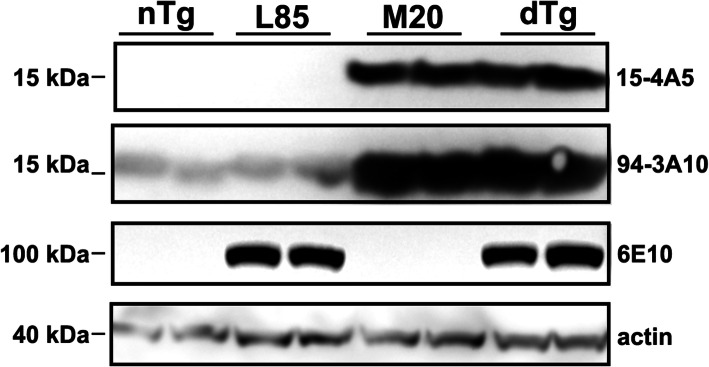
To assess the interplay between α-synuclein and Aβ on protein aggregation kinetics, we crossed mice expressing human α-synuclein (M20) with APPswe/PS1dE9 transgenic mice (L85) to generate M20/L85 mice. We then injected α-synuclein preformed fibrils (PFFs) unilaterally into the hippocampus of 6-month-old mice, harvesting 2 or 4 months later.
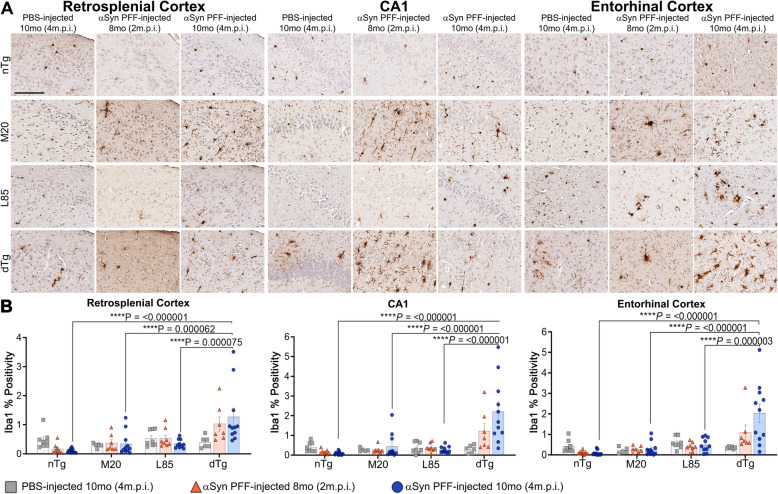
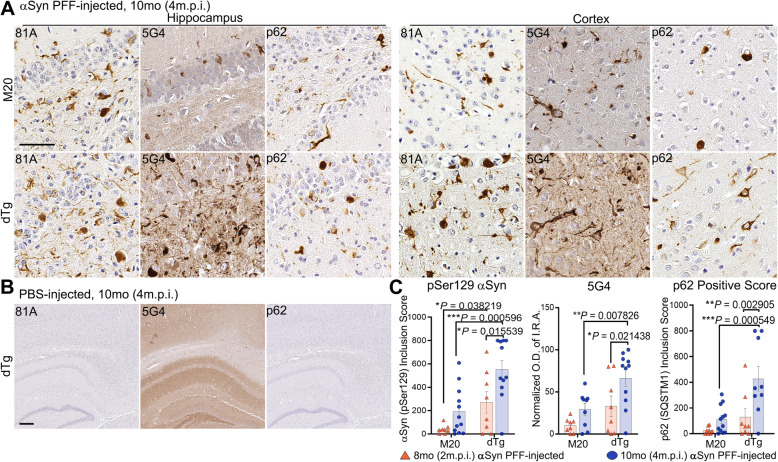
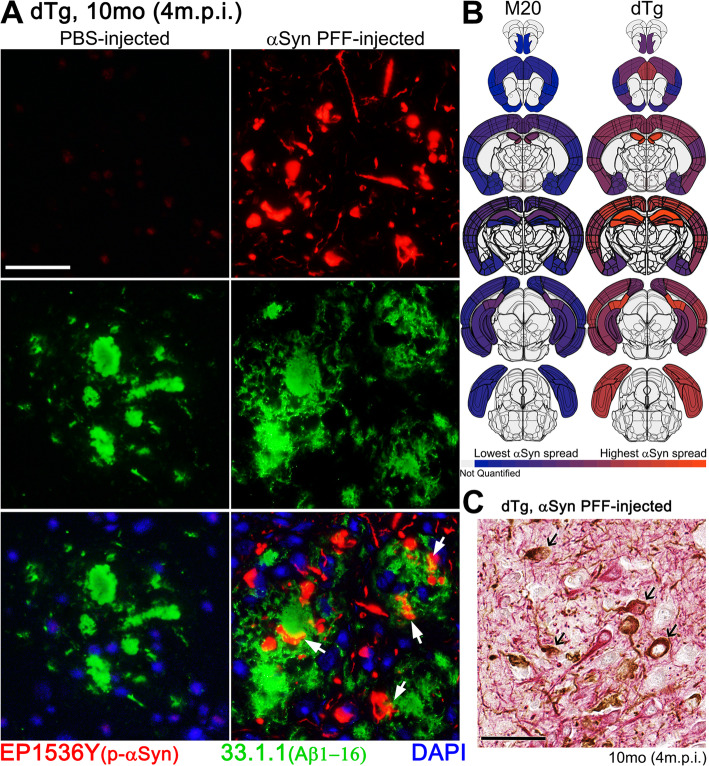
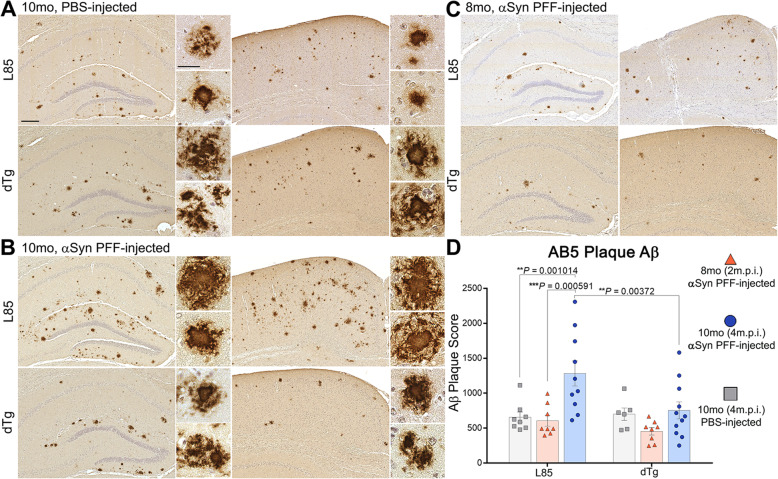
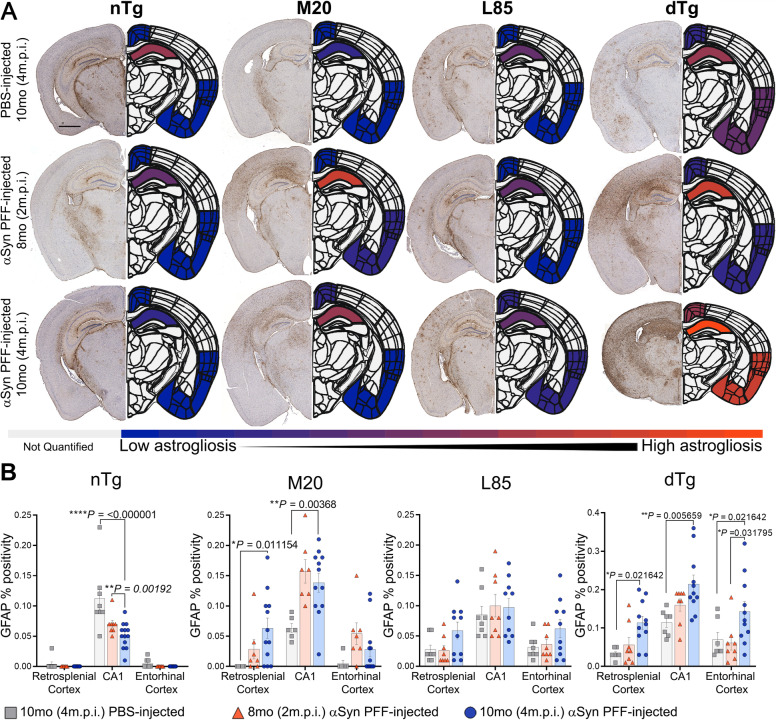
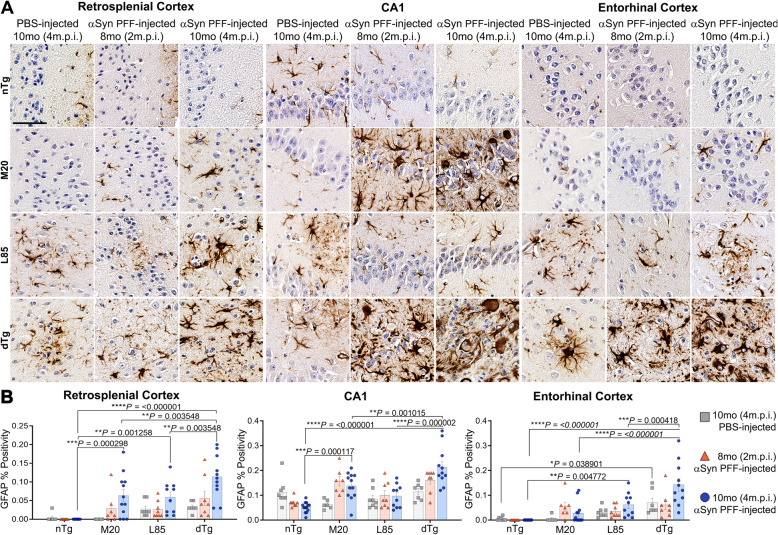
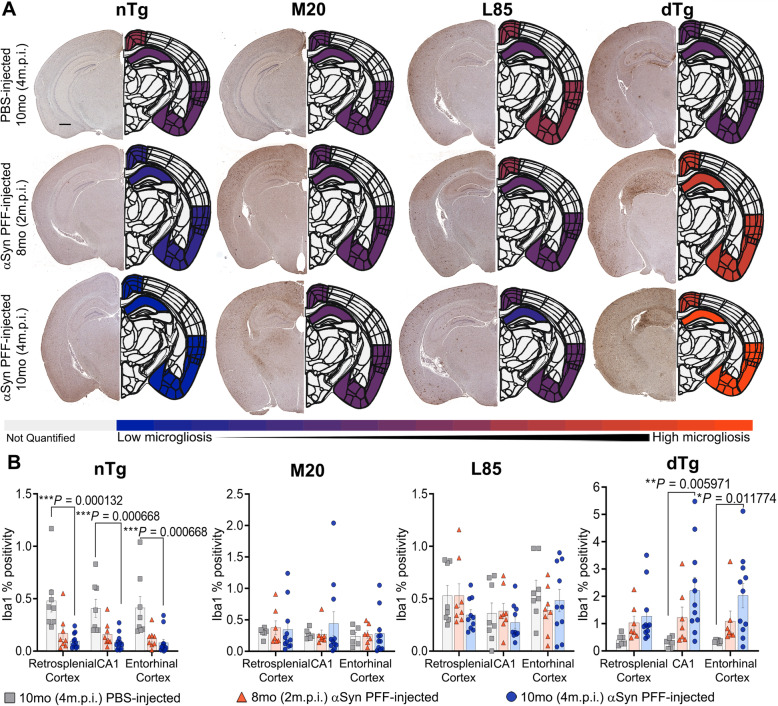
Immunohistochemical analysis of M20/L85 mice revealed that pre-existing Aβ plaques exacerbate the spread and deposition of induced α-synuclein pathology. This process was associated with increased neuroinflammation. Unexpectedly, the injection of α-synuclein PFFs in L85 mice enhanced the deposition of Aβ; whereas the level of Aβ deposition in M20/L85 bigenic mice, injected with α-synuclein PFFs, did not differ from that of mice injected with PBS.
These studies reveal novel and unexpected interplays between α-synuclein pathology, Aβ and neuroinflammation in mice that recapitulate the pathology of Alzheimer's disease and Lewy body dementia.
宿主编码蛋白错误折叠成病理性朊病毒构象是许多神经退行性疾病的一个特征,包括阿尔茨海默病、帕金森病和路易体痴呆。目前一个研究热点是这些蛋白质的病理性沉积如何相互影响,因为各种朊病毒蛋白的共病组合与疾病恶化有关。一系列病理、遗传和生化证据为这样一种观点提供了依据,即淀粉样β(Aβ)的积累可以诱导和促进α-突触核蛋白病理,从而导致神经退行性变。
为了评估α-突触核蛋白和 Aβ 之间在蛋白聚集动力学上的相互作用,我们将表达人α-突触核蛋白(M20)的小鼠与 APPswe/PS1dE9 转基因小鼠(L85)杂交,生成 M20/L85 小鼠。然后,我们将α-突触核蛋白原纤维(PFFs)单侧注射到 6 个月大的小鼠海马体中,2 或 4 个月后进行收获。
免疫组织化学分析显示,预先存在的 Aβ 斑块加剧了诱导的α-突触核蛋白病理的传播和沉积。这一过程与神经炎症的增加有关。出乎意料的是,L85 小鼠中α-突触核蛋白 PFFs 的注射增强了 Aβ的沉积;而在注射α-突触核蛋白 PFFs 的 M20/L85 双基因小鼠中,Aβ的沉积水平与注射 PBS 的小鼠没有差异。
这些研究揭示了在模拟阿尔茨海默病和路易体痴呆病理的小鼠中,α-突触核蛋白病理、Aβ 和神经炎症之间存在新的、意想不到的相互作用。