Bennedbæk Marc, Zhukova Anna, Tang Man-Hung Eric, Bennet Jaclyn, Munderi Paula, Ruxrungtham Kiat, Gisslen Magnus, Worobey Michael, Lundgren Jens D, Marvig Rasmus L
Centre of Excellence for Health, Immunity and Infection (CHIP), Department of Infectious Diseases, Rigshospitalet, University of Copenhagen, Blegdamsvej 9, 2100 Copenhagen, Denmark.
Unité Bioinformatique Evolutive, Hub Bioinformatique et Biostatistique, USR3756 (C3BI//DBC), Institut Pasteur and CNRS, 25-28 Rue du Dr Roux, 75015 Paris, France.
Virus Evol. 2021 Jun 9;7(2):veab055. doi: 10.1093/ve/veab055. eCollection 2021.
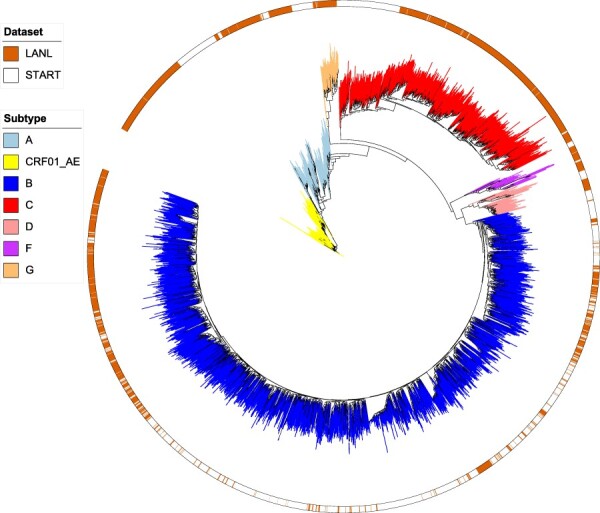
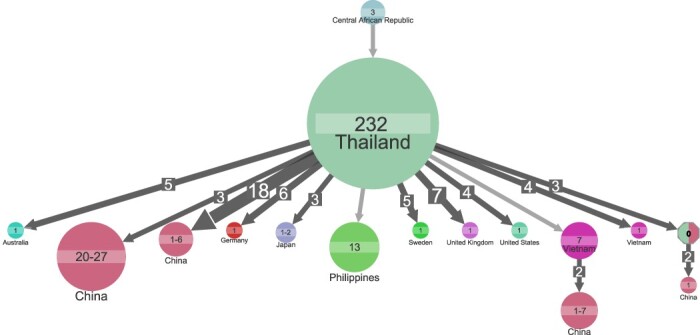
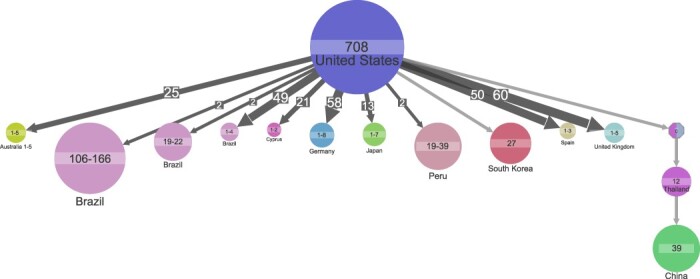
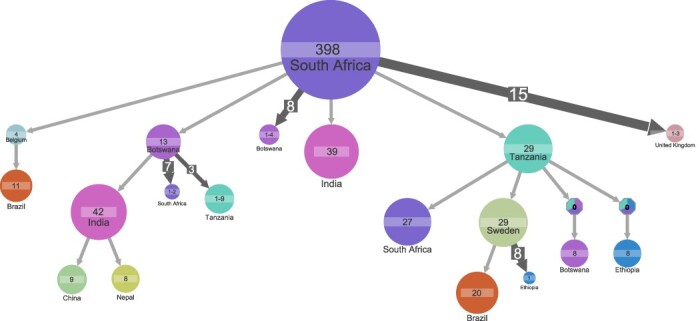
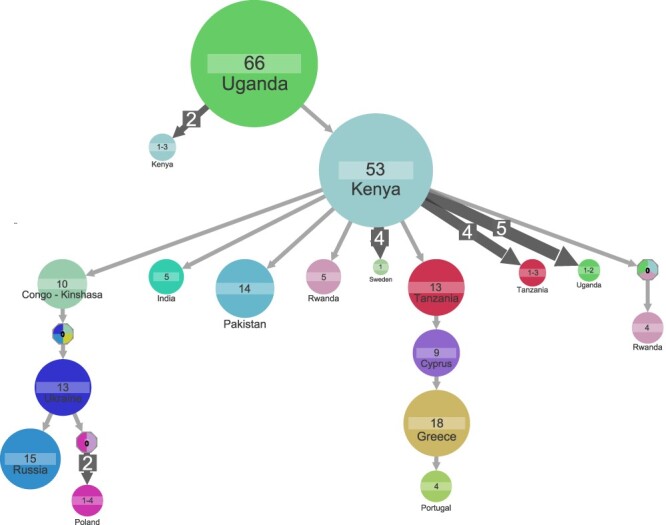
Understanding of pandemics depends on the characterization of pathogen collections from well-defined and demographically diverse cohorts. Since its emergence in Congo almost a century ago, Human Immunodeficiency Virus Type 1 (HIV-1) has geographically spread and genetically diversified into distinct viral subtypes. Phylogenetic analysis can be used to reconstruct the ancestry of the virus to better understand the origin and distribution of subtypes. We sequenced two 3.6-kb amplicons of HIV-1 genomes from 3,197 participants in a clinical trial with consistent and uniform sampling at sites across 35 countries and analyzed our data with another 2,632 genomes that comprehensively reflect the HIV-1 genetic diversity. We used maximum likelihood phylogenetic analysis coupled with geographical information to infer the state of ancestors. The majority of our sequenced genomes ( = 2,501) were either pure subtypes (A-D, F, and G) or CRF01_AE. The diversity and distribution of subtypes across geographical regions differed; USA showed the most homogenous subtype population, whereas African samples were most diverse. We delineated transmission of the four most prevalent subtypes in our dataset (A, B, C, and CRF01_AE), and our results suggest both continuous and frequent transmission of HIV-1 over country borders, as well as single transmission events being the seed of endemic population expansions. Overall, we show that coupling of genetic and geographical information of HIV-1 can be used to understand the origin and spread of pandemic pathogens.
对大流行病的理解取决于对来自明确界定且人口结构多样的队列中的病原体样本的特征描述。自近一个世纪前在刚果出现以来,1型人类免疫缺陷病毒(HIV-1)已在地理上传播并在基因上分化为不同的病毒亚型。系统发育分析可用于重建病毒的谱系,以更好地了解亚型的起源和分布。我们对一项临床试验中3197名参与者的HIV-1基因组的两个3.6 kb扩增子进行了测序,在35个国家的各个地点进行了一致且统一的采样,并将我们的数据与另外2632个全面反映HIV-1基因多样性的基因组进行了分析。我们使用最大似然系统发育分析结合地理信息来推断祖先的状态。我们测序的大多数基因组(= 2501个)要么是纯亚型(A-D、F和G),要么是CRF01_AE。亚型在地理区域的多样性和分布有所不同;美国显示出最同质化的亚型群体,而非洲样本最为多样。我们描绘了数据集中四种最流行亚型(A、B、C和CRF01_AE)的传播情况,我们的结果表明HIV-1在国家边界上持续且频繁地传播,以及单次传播事件是地方流行人群扩张的种子。总体而言,我们表明HIV-1的遗传信息和地理信息相结合可用于了解大流行病原体的起源和传播。