Wang Qi, Liu Liang, Cao Jiangang, Abula Muhetidier, Yimingjiang Yasen, Feng Shiqing
Department of Orthopedics, Tianjin Medical University General Hospital, Tianjin, China.
International Science and Technology Cooperation Base of Spinal Cord Injury, Tianjin Key Laboratory of Spine and Spinal Cord Injury, Department of Orthopedics, Tianjin Medical University General Hospital, Tianjin, China.
Ann Transl Med. 2021 Aug;9(15):1248. doi: 10.21037/atm-21-3586.
The process of spinal cord injury involves acute, subacute, and chronic stages; however, the specific pathological mechanism remains unclear. In this study, weighted gene co-expression network analysis (WGCNA) was used to clarify specific modules and hub genes that associated with SCI.
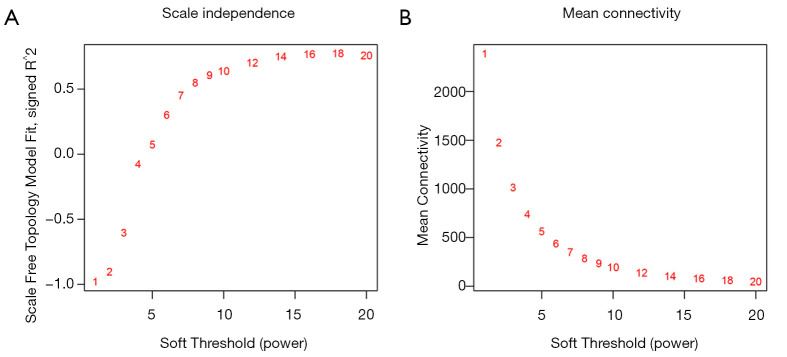
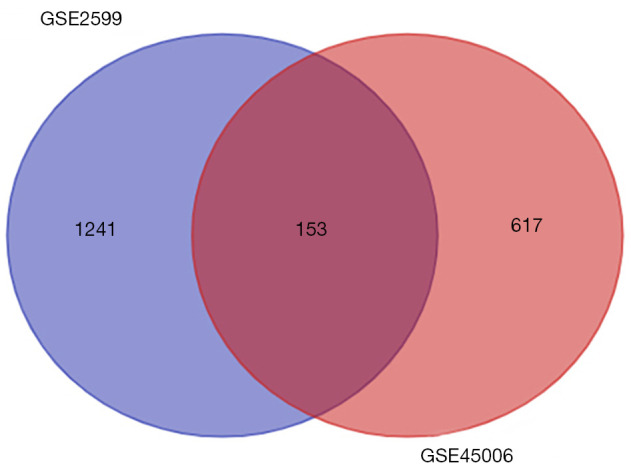
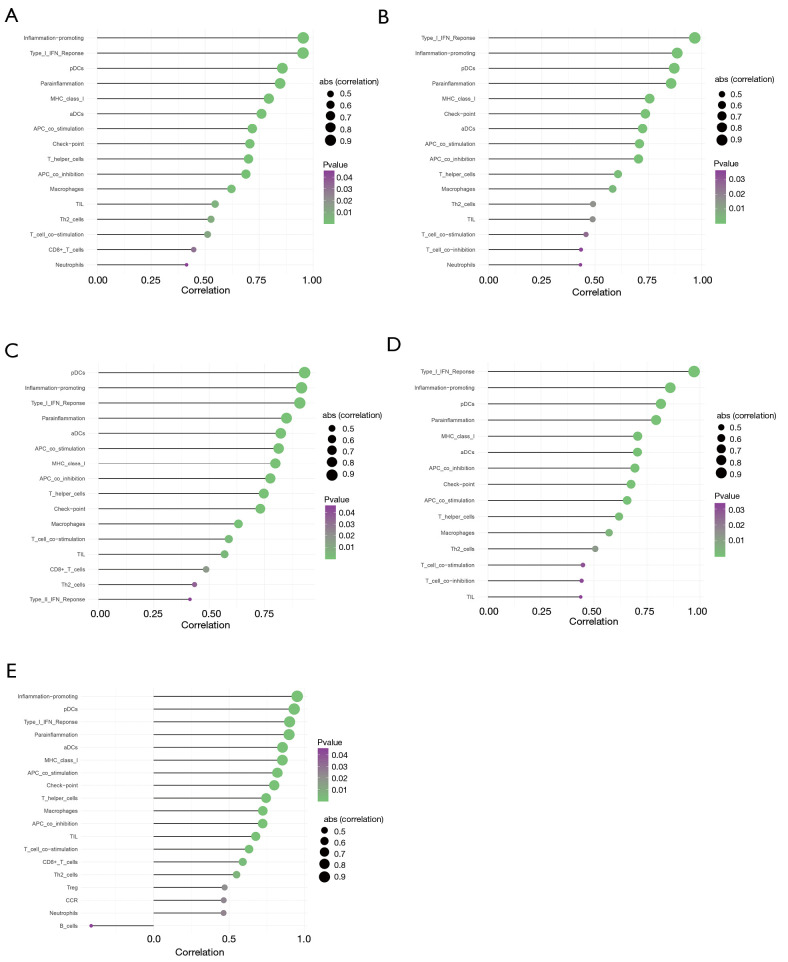
The gene expression profiles GEO Series (GSE)45006 and GEO Series (GSE)2599 were downloaded, and the co-expression network modules were identified by the WGCNA package. The protein-protein interaction (PPI) network and Venn diagram were constructed to identify hub genes. Quantitative real-time polymerase chain reaction (QRT-PCR) was used to quantify the degree of the top five candidate genes. Correlation analysis was also carried out between hub genes and immune infiltration.
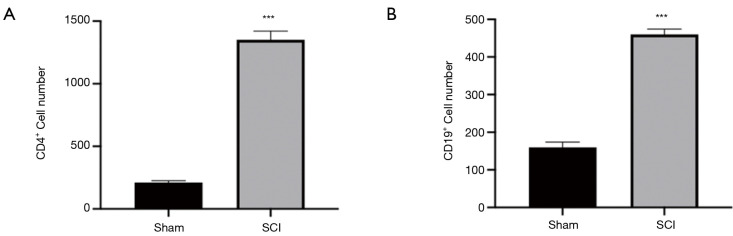
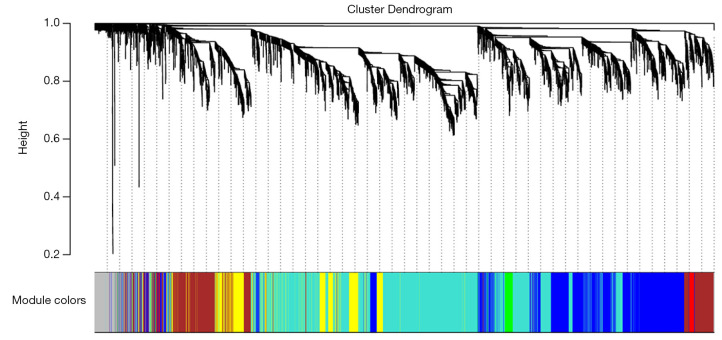
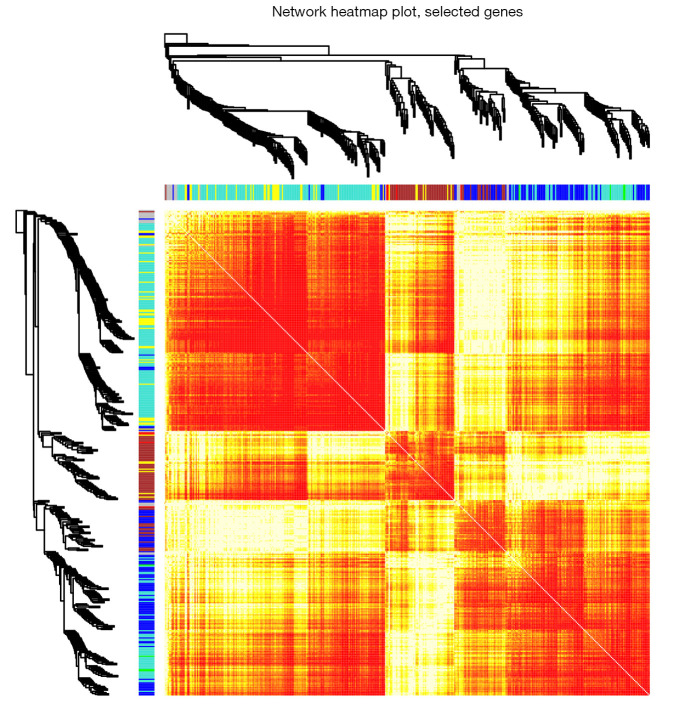
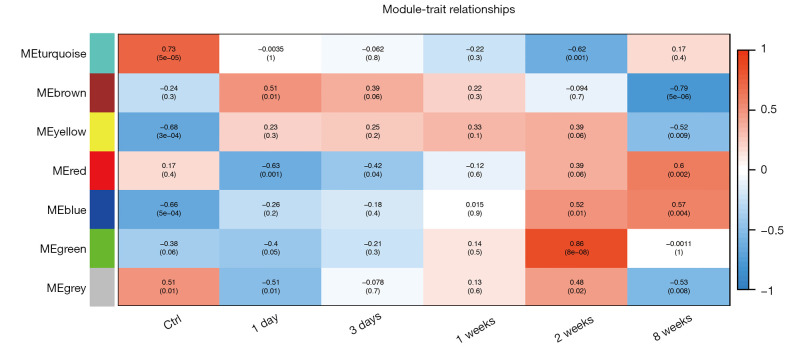
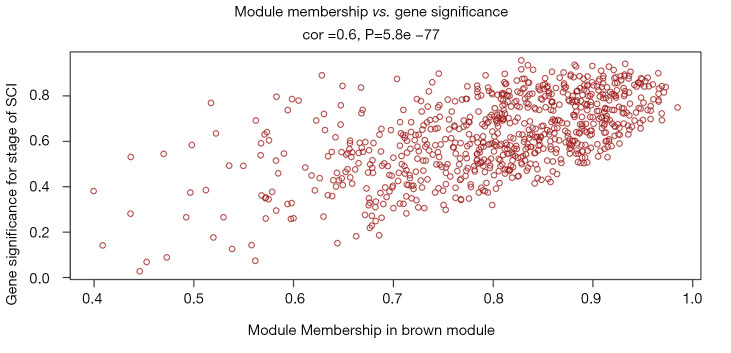
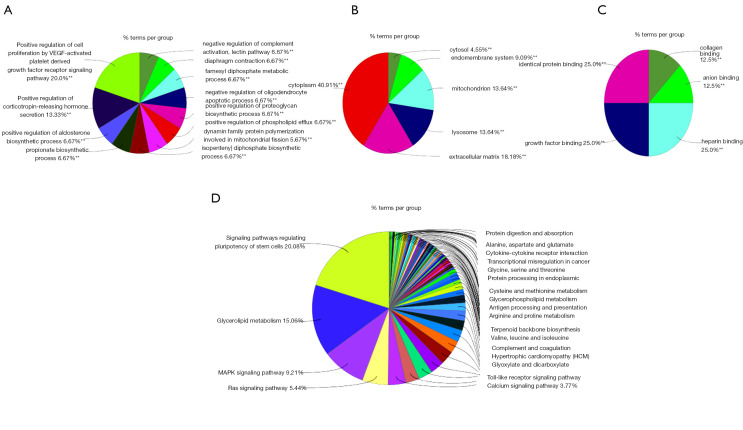
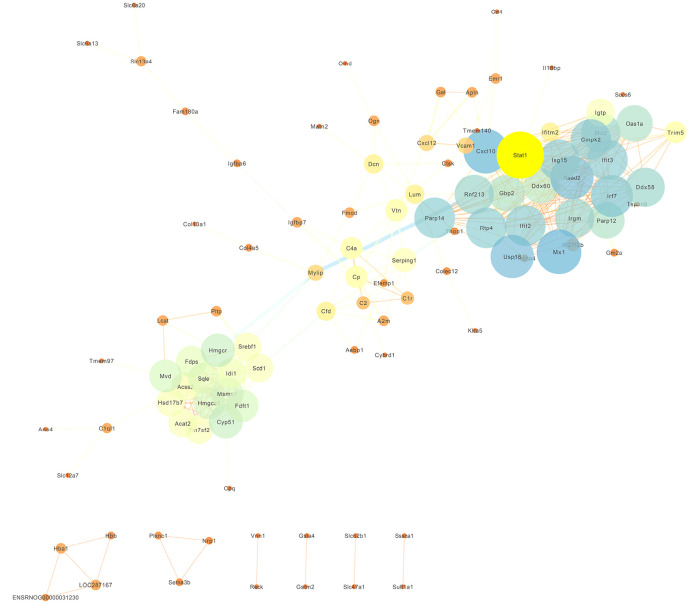
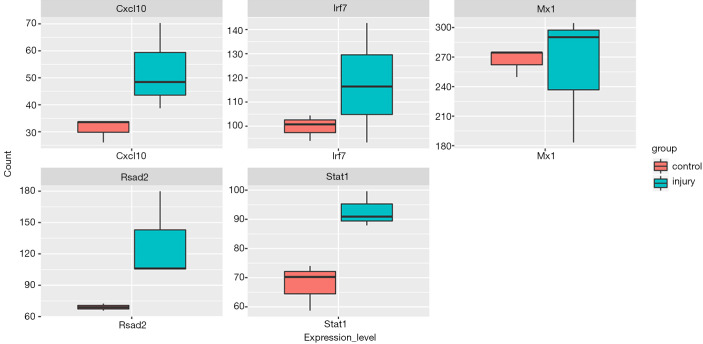
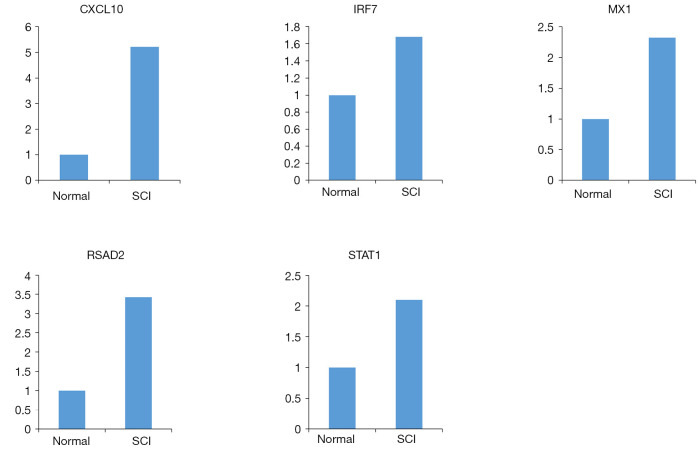
In total, 14,402 genes and seven modules were identified. The brown module was considered to be the most critical module for the chronic stage of SCI, which contained 775 genes that were primarily associated with various biological processes, including extracellular structure organization, lysosome, isoprenoid biosynthesis, response to nutrients, response to wounding, sulfur compound metabolic process, cofactor metabolic process, and ossification. Furthermore, C-X-C motif chemokine ligand 10 (), myxovirus (influenza virus) resistance 1 (), signal transducer and activator of transcription 1 (), interferon regulatory factor 7 () and radical S-adenosyl methionine domain containing 2 () were identified as the hub genes in the PPI and Venn diagram network, and verified by qRT-PCR. Immune infiltration analysis revealed that CD8+ T cells, macrophages, neutrophils, plasmacytoid dendritic cells, helper T cells, Th2 cells, and tumor-infiltrating lymphocytes may be involved in the SCI process.
There were significant differences among the five hub genes (, , , , and ) of the brown module, which may be potential diagnostic and prognostic markers of SCI, and immune cell infiltration may play an important role in the chronic stage of SCI.
脊髓损伤过程涉及急性、亚急性和慢性阶段;然而,具体的病理机制仍不清楚。在本研究中,采用加权基因共表达网络分析(WGCNA)来阐明与脊髓损伤相关的特定模块和枢纽基因。
下载基因表达谱GEO系列(GSE)45006和GEO系列(GSE)2599,并通过WGCNA软件包识别共表达网络模块。构建蛋白质-蛋白质相互作用(PPI)网络和维恩图以识别枢纽基因。采用定量实时聚合酶链反应(QRT-PCR)对排名前五的候选基因的表达程度进行定量。还对枢纽基因与免疫浸润之间进行了相关性分析。
共鉴定出14402个基因和7个模块。棕色模块被认为是脊髓损伤慢性期最关键的模块,其中包含775个基因,这些基因主要与各种生物学过程相关,包括细胞外结构组织、溶酶体、类异戊二烯生物合成、对营养物质的反应、对损伤的反应、硫化合物代谢过程、辅因子代谢过程和骨化。此外,C-X-C基序趋化因子配体10(CXCL10)、黏液病毒(流感病毒)抗性1(MX1)、信号转导和转录激活因子1(STAT1)、干扰素调节因子7(IRF7)和含自由基S-腺苷甲硫氨酸结构域2(RSAD2)在PPI和维恩图网络中被鉴定为枢纽基因,并通过qRT-PCR进行了验证。免疫浸润分析显示,CD8 + T细胞、巨噬细胞、中性粒细胞、浆细胞样树突状细胞、辅助性T细胞、Th2细胞和肿瘤浸润淋巴细胞可能参与脊髓损伤过程。
棕色模块的五个枢纽基因(CXCL10、MX1、STAT1、IRF7和RSAD2)存在显著差异,可能是脊髓损伤潜在的诊断和预后标志物,免疫细胞浸润可能在脊髓损伤慢性期发挥重要作用。