Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), Inserm U 1258, CNRS UMR 7104, Université de Strasbourg, Illkirch, France.
Laboratoire de Diagnostic Génétique, Faculté de Médecine, CHRU, Strasbourg, France.
Acta Neuropathol Commun. 2021 Sep 17;9(1):155. doi: 10.1186/s40478-021-01254-y.
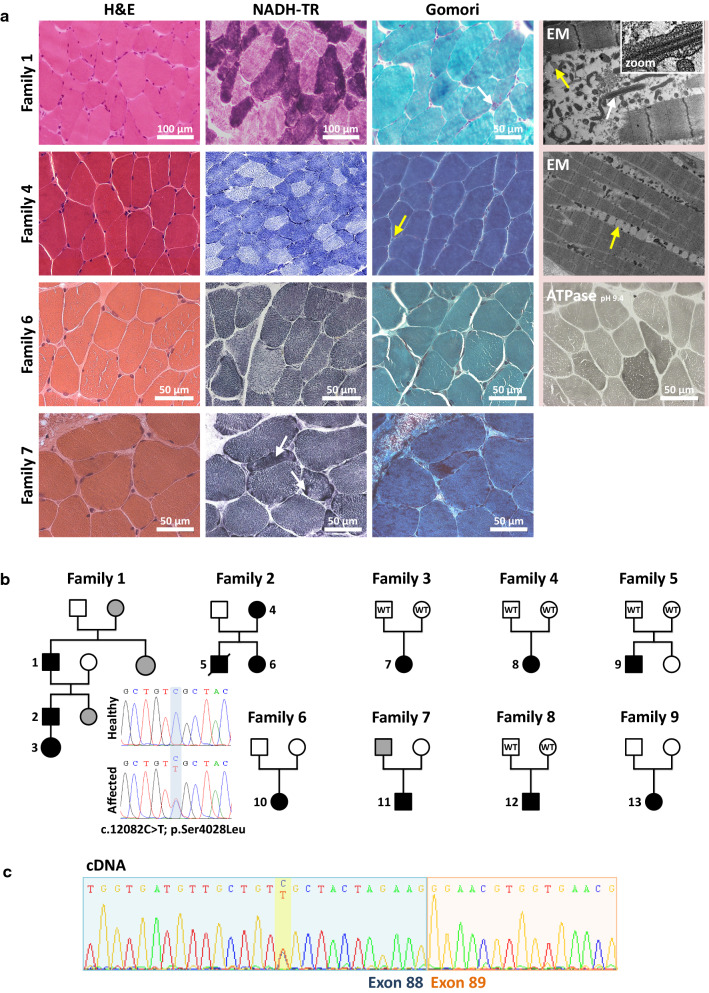
The ryanodine receptor RyR1 is the main sarcoplasmic reticulum Ca channel in skeletal muscle and acts as a connecting link between electrical stimulation and Ca-dependent muscle contraction. Abnormal RyR1 activity compromises normal muscle function and results in various human disorders including malignant hyperthermia, central core disease, and centronuclear myopathy. However, RYR1 is one of the largest genes of the human genome and accumulates numerous missense variants of uncertain significance (VUS), precluding an efficient molecular diagnosis for many patients and families. Here we describe a recurrent RYR1 mutation previously classified as VUS, and we provide clinical, histological, and genetic data supporting its pathogenicity. The heterozygous c.12083C>T (p.Ser4028Leu) mutation was found in thirteen patients from nine unrelated congenital myopathy families with consistent clinical presentation, and either segregated with the disease in the dominant families or occurred de novo. The affected individuals essentially manifested neonatal or infancy-onset hypotonia, delayed motor milestones, and a benign disease course differing from classical RYR1-related muscle disorders. Muscle biopsies showed unspecific histological and ultrastructural findings, while RYR1-typical cores and internal nuclei were seen only in single patients. In conclusion, our data evidence the causality of the RYR1 c.12083C>T (p.Ser4028Leu) mutation in the development of an atypical congenital myopathy with gradually improving motor function over the first decades of life, and may direct molecular diagnosis for patients with comparable clinical presentation and unspecific histopathological features on the muscle biopsy.
兰尼碱受体 RyR1 是骨骼肌肌浆网中的主要钙通道,它是电刺激和 Ca 依赖性肌肉收缩之间的连接环节。RyR1 活性异常会损害正常的肌肉功能,并导致各种人类疾病,包括恶性高热、中央核疾病和中核肌病。然而,RYR1 是人类基因组中最大的基因之一,积累了大量意义不明的错义变异(VUS),这使得许多患者和家庭无法进行有效的分子诊断。在这里,我们描述了一个先前被归类为 VUS 的 RYR1 突变,并提供了支持其致病性的临床、组织学和遗传数据。杂合子 c.12083C>T(p.Ser4028Leu)突变在九个无关先天性肌病家族的 13 名患者中发现,具有一致的临床表现,在显性家族中与疾病共分离,或为新发突变。受影响的个体主要表现为新生儿或婴儿期起病的肌无力、运动发育迟缓,以及良性的疾病过程,与经典的 RYR1 相关肌肉疾病不同。肌肉活检显示出非特异性的组织学和超微结构表现,而 RYR1 典型的核心和内核仅在单个患者中观察到。总之,我们的数据证明了 RYR1 c.12083C>T(p.Ser4028Leu)突变在发展一种非典型先天性肌病中的因果关系,这种肌病在生命的最初几十年中运动功能逐渐改善,并且可能为具有类似临床表现和非特异性组织病理学特征的患者提供分子诊断。