Department of Food Science, Cornell Universitygrid.5386.8, Ithaca, New York, USA.
mSphere. 2021 Oct 27;6(5):e0048521. doi: 10.1128/mSphere.00485-21. Epub 2021 Sep 22.
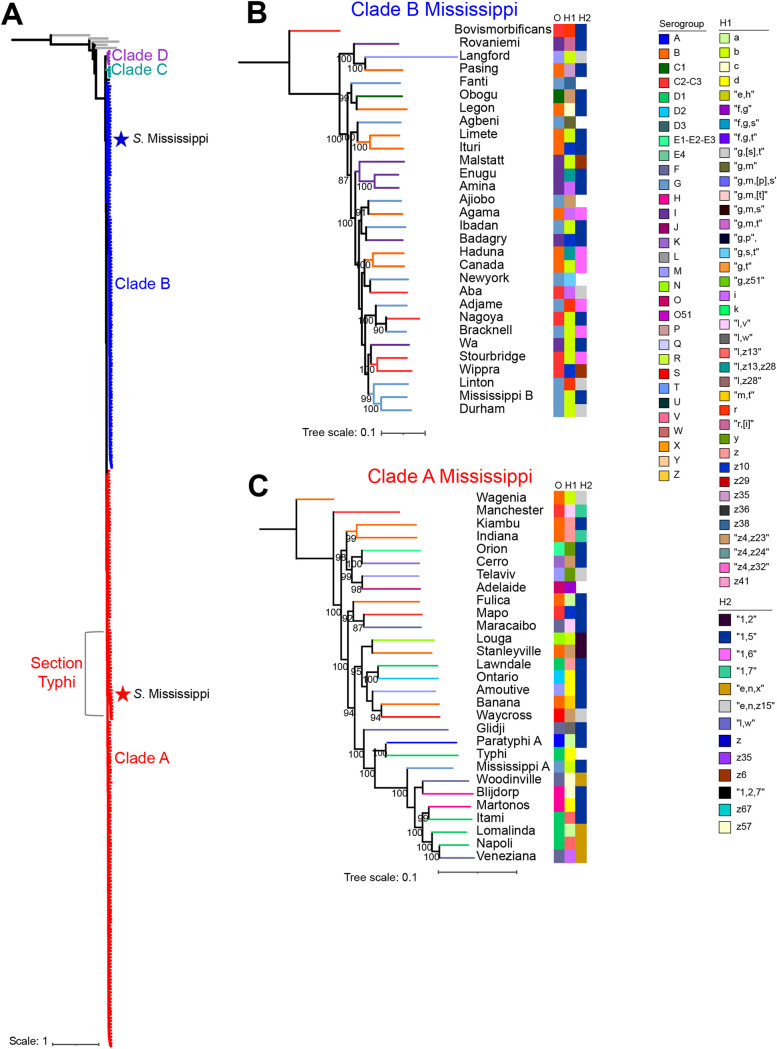
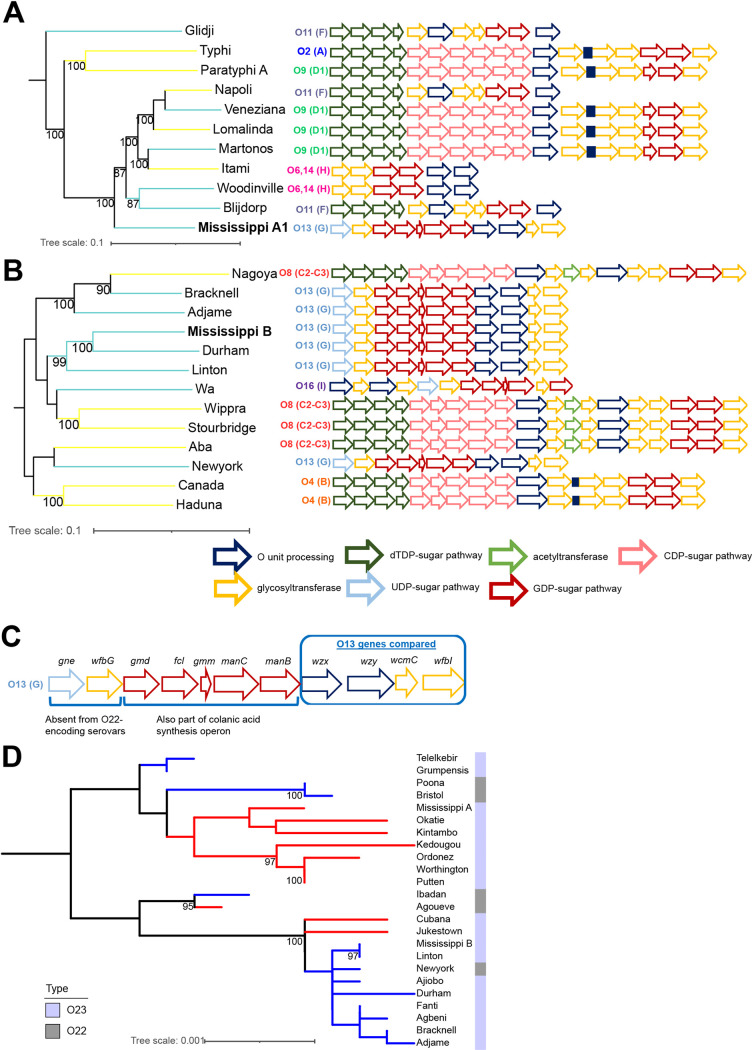
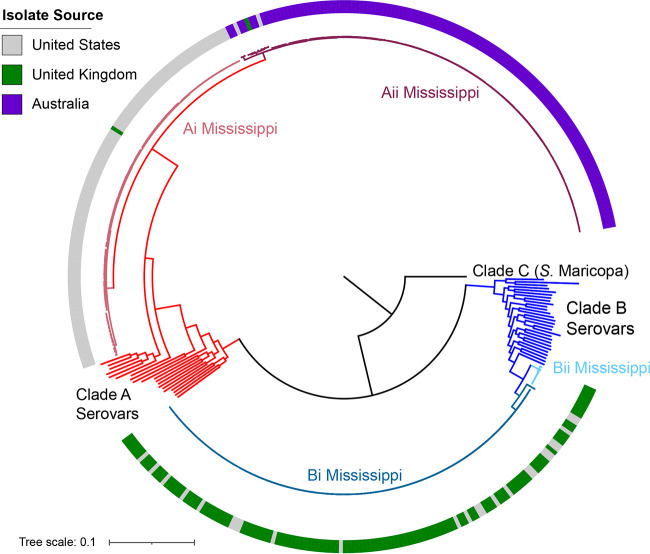
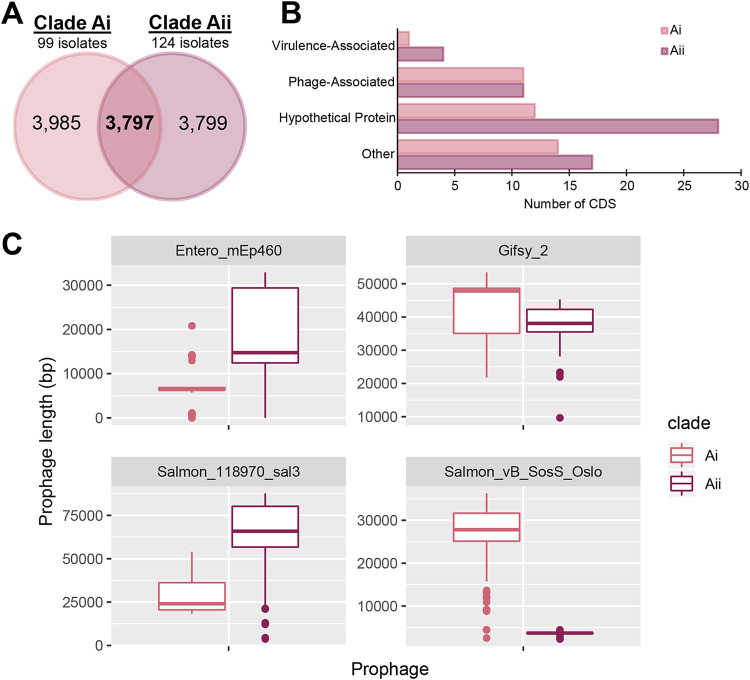
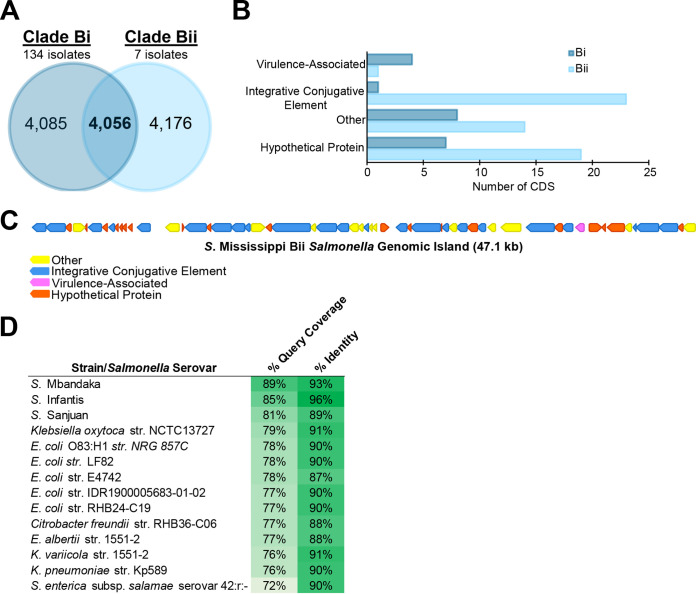
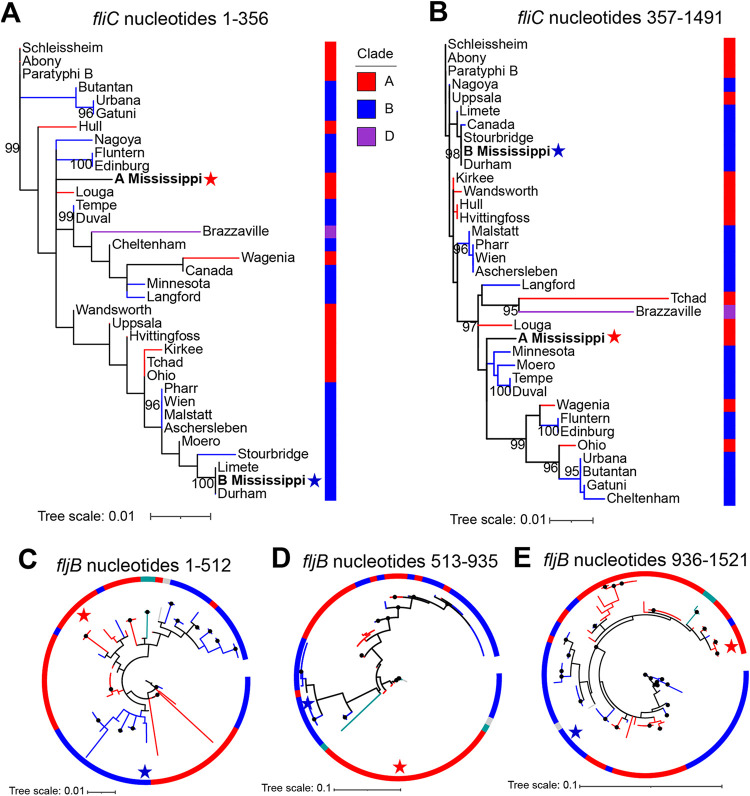
Salmonella enterica serovar Mississippi is the 2nd and 14th leading cause of human clinical salmonellosis in the Australian island state of Tasmania and the United States, respectively. Despite its public health relevance, relatively little is known about this serovar. Comparison of whole-genome sequence (WGS) data of Mississippi isolates with WGS data for 317 additional S. enterica serovars placed one clade of Mississippi within S. enterica clade B ("clade B Mississippi") and the other within section Typhi in S. enterica clade A ("clade A Mississippi"), suggesting that these clades evolved from different ancestors. Phylogenetic analysis of 364 Mississippi isolates from Australia, the United Kingdom, and the United States suggested that the isolates cluster geographically, with U.S. and Australian isolates representing different subclades (Ai and Aii, respectively) within clade A Mississippi and clade B isolates representing the predominant Mississippi isolates in the United Kingdom. Intraclade comparisons suggested that different mobile elements, some of which encode virulence factors, are responsible for the observed differences in gene content among isolates within these clades. Specifically, genetic differences among clade A isolates reflect differences in prophage contents, while differences among clade B isolates are due to the acquisition of a 47.1-kb integrative conjugative element (ICE). Phylogenies inferred from antigenic components (, , and O-antigen-processing genes) support that clade A and B Mississippi isolates acquired these loci from different ancestral serovars. Overall, these data support that different Mississippi phylogenetic clades are endemic in Australia, the United Kingdom, and the United States. The number of known so-called "polyphyletic" serovars (i.e., phylogenetically distinct clades with the same O and H antigenic formulas) continues to increase as additional Salmonella isolates are sequenced. While serotyping remains a valuable tool for reporting and monitoring Salmonella, more discriminatory analyses for classifying polyphyletic serovars may improve surveillance efforts for these serovars, as we found that for Mississippi, distinct genotypes predominate at different geographic locations. Our results suggest that the acquisition of genes encoding O and H antigens from different ancestors led to the emergence of two Mississippi clades. Furthermore, our results suggest that different mobile elements contribute to the microevolution and diversification of isolates within these two clades, which has implications for the acquisition of novel adaptations, such as virulence factors.
肠炎沙门氏菌密西西比血清型分别是澳大利亚塔斯马尼亚州和美国导致人类临床沙门氏菌病的第 2 和第 14 大原因。尽管其具有公共卫生相关性,但人们对该血清型的了解相对较少。将密西西比分离株的全基因组序列 (WGS) 数据与 317 种其他肠炎沙门氏菌血清型的 WGS 数据进行比较,将一个密西西比分枝置于肠炎沙门氏菌 B 分枝(“B 分枝密西西比”)内,另一个分枝置于肠炎沙门氏菌 A 分枝的伤寒血清型内(“A 分枝密西西比”),表明这些分枝是由不同的祖先进化而来。对来自澳大利亚、英国和美国的 364 株密西西比分离株的系统发育分析表明,这些分离株在地理上聚类,美国和澳大利亚的分离株分别代表 A 分枝密西西比内的不同亚分枝(Ai 和 Aii),而 B 分枝分离株则代表英国主要的密西西比分离株。在分枝内比较表明,不同的移动元件(其中一些编码毒力因子)是导致这些分枝内分离株基因含量存在差异的原因。具体而言,A 分枝分离株之间的遗传差异反映了噬菌体含量的差异,而 B 分枝分离株之间的差异则归因于一个 47.1 kb 的整合性接合元件(ICE)的获得。基于抗原成分(,和 O-抗原加工基因)推断的系统发育表明,A 分枝和 B 分枝的密西西比分离株从不同的祖先血清型获得了这些基因座。总体而言,这些数据支持澳大利亚、英国和美国存在不同的密西西比分支。随着更多沙门氏菌分离株的测序,已知所谓的“多系”(即具有相同 O 和 H 抗原公式但系统发育不同的分枝)的数量继续增加。虽然血清分型仍然是报告和监测沙门氏菌的一种有价值的工具,但更具区分性的分析方法可用于分类多系血清型,以改善对这些血清型的监测工作,因为我们发现对于密西西比,不同的基因型在不同的地理位置占主导地位。我们的研究结果表明,从不同祖先获得编码 O 和 H 抗原的基因导致了两个密西西比分枝的出现。此外,我们的研究结果表明,不同的移动元件导致这些分枝内分离株的微进化和多样化,这对新适应的获得(如毒力因子)有影响。