Joint Institute for Food Safety and Applied Nutrition, University of Maryland, College Park, Maryland, USA
Joint Institute for Food Safety and Applied Nutrition, University of Maryland, College Park, Maryland, USA.
mBio. 2018 Nov 27;9(6):e02303-18. doi: 10.1128/mBio.02303-18.
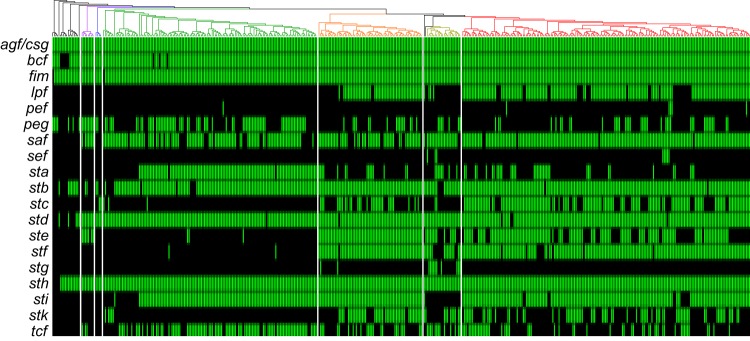
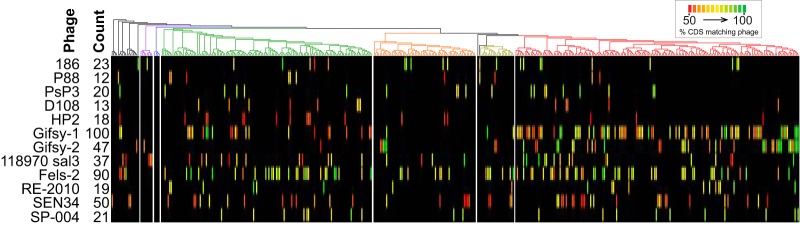
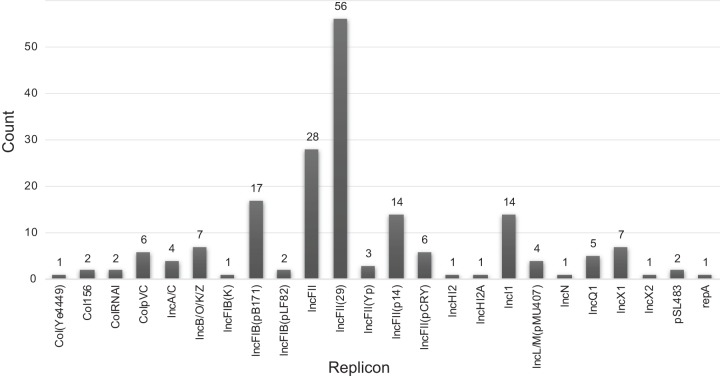
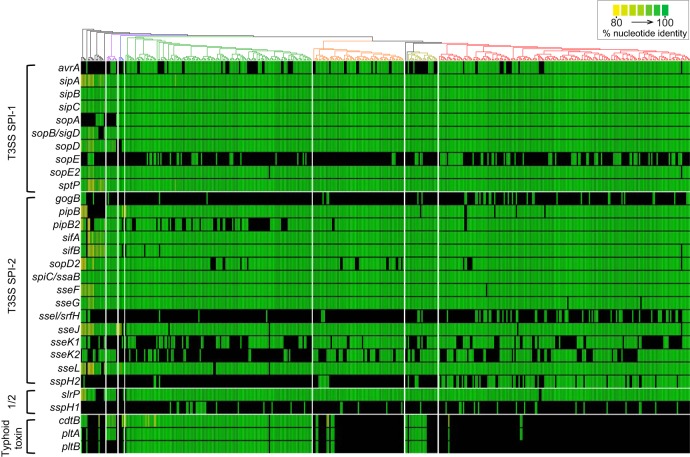
Using whole-genome sequence (WGS) data from the GenomeTrakr network, a globally distributed network of laboratories sequencing foodborne pathogens, we present a new phylogeny of comprising 445 isolates from 266 distinct serovars and originating from 52 countries. This phylogeny includes two previously unidentified subsp. clades. Serovar Typhi is shown to be nested within clade A. Our findings are supported by both phylogenetic support, based on a core genome alignment, and Bayesian approaches, based on single-nucleotide polymorphisms. Serovar assignments were refined by analysis using SeqSero. More than 10% of serovars were either polyphyletic or paraphyletic. We found variable genetic content in these isolates relating to gene mobilization and virulence factors which have different distributions within clades. Gifsy-1- and Gifsy-2-like phages appear more prevalent in clade A; other viruses are more evenly distributed. Our analyses reveal IncFII is the predominant plasmid replicon in Few core or clade-defining virulence genes are observed, and their distributions appear probabilistic in nature. Together, these patterns demonstrate that genetic exchange within is more extensive and frequent than previously realized, which significantly alters how we view the genetic structure of the bacterial species. Rapid improvements in nucleotide sequencing access and affordability have led to a drastic increase in availability of genetic information. This information will improve the accuracy of molecular descriptions, including serovars, within Although the concept of serovars continues to be useful, it may have more significant limitations than previously understood. Furthermore, the discrete absence or presence of specific genes can be an unstable indicator of phylogenetic identity. Whole-genome sequencing provides more rigorous tools for assessing the distributions of these genes. Our phylogenetic and genetic content analyses reveal how active genetic elements are dynamically distributed within a species, allowing us to better understand genetic reservoirs and underlying bacterial evolution.
利用全球分布的实验室测序食源性病原体的 GenomeTrakr 网络的全基因组序列 (WGS) 数据,我们呈现了一个由来自 52 个国家的 266 个不同血清型的 445 个分离株组成的新系统发育。该系统发育包括两个以前未识别的 亚种。副伤寒血清型 Typhi 被嵌套在 A 群中。我们的发现得到了核心基因组比对的基于系统发育支持和单核苷酸多态性的贝叶斯方法的支持。使用 SeqSero 进行 分析来细化血清型分配。超过 10%的血清型是多源的或并系的。我们在这些分离株中发现了与基因转移和毒力因子有关的可变遗传物质,这些因子在不同的分支中分布不同。Gifsy-1-和 Gifsy-2 样噬菌体在 A 群中更为普遍;其他病毒分布更为均匀。我们的分析表明 IncFII 是 中主要的质粒复制子。很少观察到核心或分支定义的毒力基因,它们的分布似乎是概率性质的。这些模式表明, 内的基因交换比以前认识到的更为广泛和频繁,这极大地改变了我们对细菌物种遗传结构的看法。核苷酸测序获取和负担能力的快速提高导致遗传信息的可用性急剧增加。这些信息将提高包括血清型在内的分子描述的准确性。虽然血清型的概念仍然有用,但它可能比以前理解的具有更大的局限性。此外,特定基因的缺失或存在与否可能是系统发育身份的不稳定指标。全基因组测序为评估这些基因的分布提供了更严格的工具。我们的系统发育和遗传内容分析揭示了活跃的遗传元素如何在一个物种内动态分布,使我们能够更好地了解遗传库和潜在的细菌进化。