Department of Biochemistry and Biophysics, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA.
Department of Medicine, Aab Cardiovascular Research Institute, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA.
Cardiovasc Res. 2022 Sep 20;118(12):2703-2717. doi: 10.1093/cvr/cvab304.
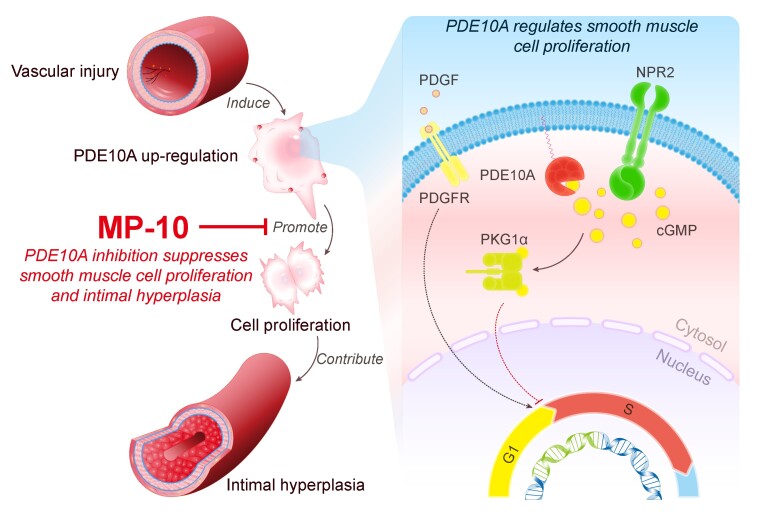
Intimal hyperplasia is a common feature of vascular remodelling disorders. Accumulation of synthetic smooth muscle cell (SMC)-like cells is the main underlying cause. Current therapeutic approaches including drug-eluting stents are not perfect due to the toxicity on endothelial cells and novel therapeutic strategies are needed. Our preliminary screening for dysregulated cyclic nucleotide phosphodiesterases (PDEs) in growing SMCs revealed the alteration of PDE10A expression. Herein, we investigated the function of PDE10A in SMC proliferation and intimal hyperplasia both in vitro and in vivo.
RT-qPCR, immunoblot, and in situ proximity ligation assay were performed to determine PDE10A expression in synthetic SMCs and injured vessels. We found that PDE10A mRNA and/or protein levels are up-regulated in cultured SMCs upon growth stimulation, as well as in intimal cells in injured mouse femoral arteries. To determine the cellular functions of PDE10A, we focused on its role in SMC proliferation. The anti-mitogenic effects of PDE10A on SMCs were evaluated via cell counting, BrdU incorporation, and flow cytometry. We found that PDE10A deficiency or inhibition arrested the SMC cell cycle at G1-phase with a reduction of cyclin D1. The anti-mitotic effect of PDE10A inhibition was dependent on cGMP-dependent protein kinase Iα (PKGIα), involving C-natriuretic peptide (CNP) and particulate guanylate cyclase natriuretic peptide receptor 2 (NPR2). In addition, the effects of genetic depletion and pharmacological inhibition of PDE10A on neointimal formation were examined in a mouse model of femoral artery wire injury. Both PDE10A knockout and inhibition decreased injury-induced intimal thickening in femoral arteries by at least 50%. Moreover, PDE10A inhibition decreased ex vivo remodelling of cultured human saphenous vein segments.
Our findings indicate that PDE10A contributes to SMC proliferation and intimal hyperplasia at least partially via antagonizing CNP/NPR2/cGMP/PKG1α signalling and suggest that PDE10A may be a novel drug target for treating vascular occlusive disease.
内膜增生是血管重构障碍的常见特征。合成平滑肌细胞(SMC)样细胞的积累是主要的潜在原因。由于对内皮细胞的毒性,目前包括药物洗脱支架在内的治疗方法并不完善,因此需要新的治疗策略。我们对生长中的 SMC 中失调的环核苷酸磷酸二酯酶(PDE)的初步筛选显示 PDE10A 表达的改变。在此,我们研究了 PDE10A 在体外和体内对 SMC 增殖和内膜增生的作用。
通过 RT-qPCR、免疫印迹和原位邻近连接测定来确定合成 SMC 和损伤血管中 PDE10A 的表达。我们发现,在生长刺激下,培养的 SMC 中 PDE10A mRNA 和/或蛋白水平上调,在损伤的小鼠股动脉内膜细胞中也是如此。为了确定 PDE10A 的细胞功能,我们重点研究了它在 SMC 增殖中的作用。通过细胞计数、BrdU 掺入和流式细胞术评估 PDE10A 对 SMC 的抗有丝分裂作用。我们发现,PDE10A 缺乏或抑制使 SMC 细胞周期在 G1 期停滞,cyclin D1 减少。PDE10A 抑制的抗有丝分裂作用依赖于 cGMP 依赖性蛋白激酶 Iα(PKGIα),涉及 C 型利钠肽(CNP)和颗粒型鸟苷酸环化酶利钠肽受体 2(NPR2)。此外,还在小鼠股动脉线损伤模型中研究了 PDE10A 的基因缺失和药理学抑制对新生内膜形成的影响。PDE10A 敲除和抑制均可使股动脉损伤引起的内膜增厚减少至少 50%。此外,PDE10A 抑制减少了培养的人隐静脉段的体外重塑。
我们的研究结果表明,PDE10A 通过拮抗 CNP/NPR2/cGMP/PKG1α 信号通路至少部分促进 SMC 增殖和内膜增生,并表明 PDE10A 可能是治疗血管闭塞性疾病的新药物靶点。