Department of Computer Science, Rice University, Houston, TX 77251-1892, USA.
Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA.
Gigascience. 2021 Sep 24;10(9). doi: 10.1093/gigascience/giab063.
Long-read sequencing has enabled unprecedented surveys of structural variation across the entire human genome. To maximize the potential of long-read sequencing in this context, novel mapping methods have emerged that have primarily focused on either speed or accuracy. Various heuristics and scoring schemas have been implemented in widely used read mappers (minimap2 and NGMLR) to optimize for speed or accuracy, which have variable performance across different genomic regions and for specific structural variants. Our hypothesis is that constraining read mapping to the use of a single gap penalty across distinct mutational hot spots reduces read alignment accuracy and impedes structural variant detection.
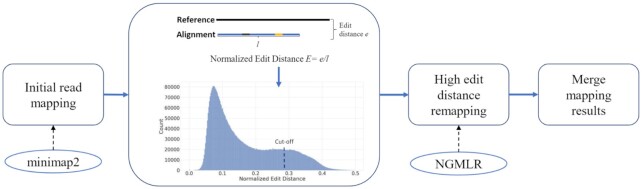
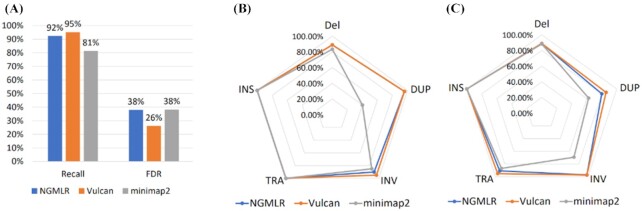
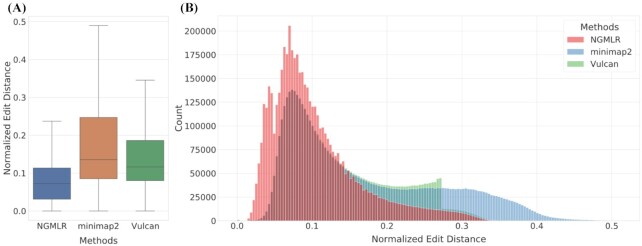
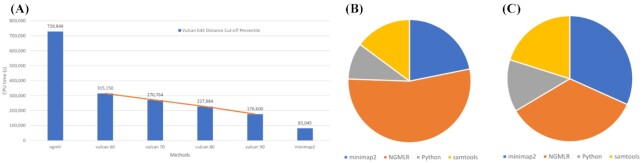
We tested our hypothesis by implementing a read-mapping pipeline called Vulcan that uses two distinct gap penalty modes, which we refer to as dual-mode alignment. The high-level idea is that Vulcan leverages the computed normalized edit distance of the mapped reads via minimap2 to identify poorly aligned reads and realigns them using the more accurate yet computationally more expensive long-read mapper (NGMLR). In support of our hypothesis, we show that Vulcan improves the alignments for Oxford Nanopore Technology long reads for both simulated and real datasets. These improvements, in turn, lead to improved accuracy for structural variant calling performance on human genome datasets compared to either of the read-mapping methods alone.
Vulcan is the first long-read mapping framework that combines two distinct gap penalty modes for improved structural variant recall and precision. Vulcan is open-source and available under the MIT License at https://gitlab.com/treangenlab/vulcan.
长读测序技术使对人类全基因组结构变异的全面调查成为可能。为了在这种情况下最大限度地发挥长读测序的潜力,出现了一些新的映射方法,这些方法主要侧重于速度或准确性。在广泛使用的读映射器(minimap2 和 NGMLR)中实现了各种启发式和评分方案,以优化速度或准确性,这些方案在不同的基因组区域和特定的结构变体上的性能各不相同。我们的假设是,将读映射约束为在不同突变热点使用单一缺口罚分,会降低读对齐的准确性,并阻碍结构变体的检测。
我们通过实现一个名为 Vulcan 的读映射管道来测试我们的假设,该管道使用两种不同的缺口罚分模式,我们称之为双模对齐。其基本思想是,Vulcan 通过 minimap2 利用映射读取的计算归一化编辑距离来识别对齐不良的读取,并使用更准确但计算成本更高的长读取映射器(NGMLR)重新对齐它们。为了支持我们的假设,我们表明,Vulcan 提高了牛津纳米孔技术长读取在模拟和真实数据集上的对齐度。这些改进反过来又提高了人类基因组数据集上结构变体调用性能的准确性,优于单独使用任何一种读映射方法。
Vulcan 是第一个结合两种不同缺口罚分模式以提高结构变体召回率和精度的长读映射框架。Vulcan 是一个开源框架,可在 MIT 许可证下通过以下网址获得:https://gitlab.com/treangenlab/vulcan。