Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, USA.
Center for Integrative Bioinformatics Vienna, Max F. Perutz Laboratories, University of Vienna, Medical University of Vienna, Vienna, Austria.
Nat Methods. 2018 Jun;15(6):461-468. doi: 10.1038/s41592-018-0001-7. Epub 2018 Apr 30.
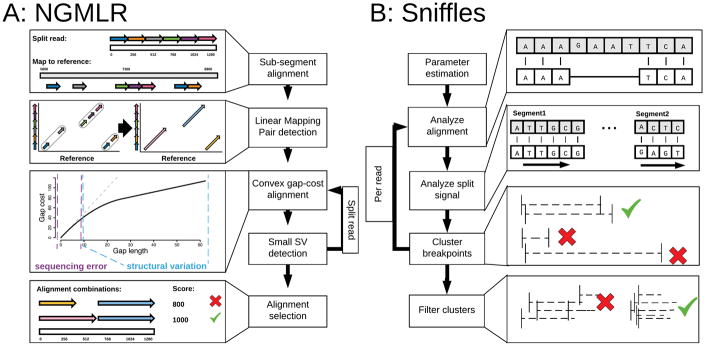
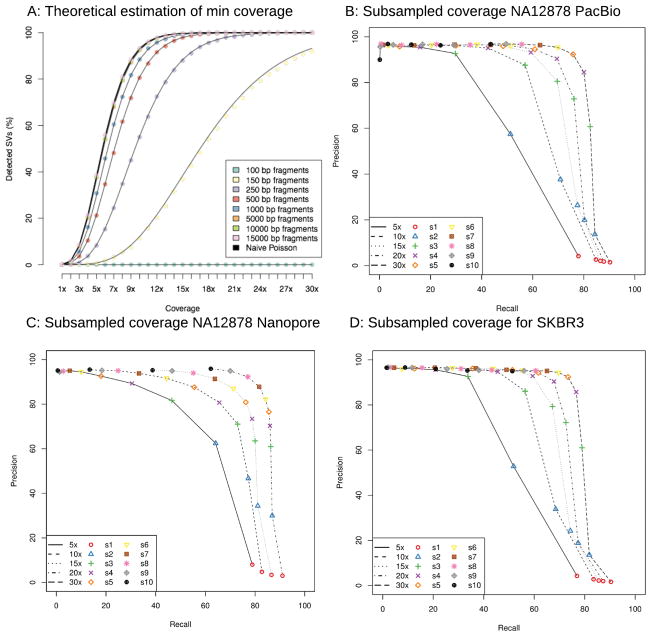
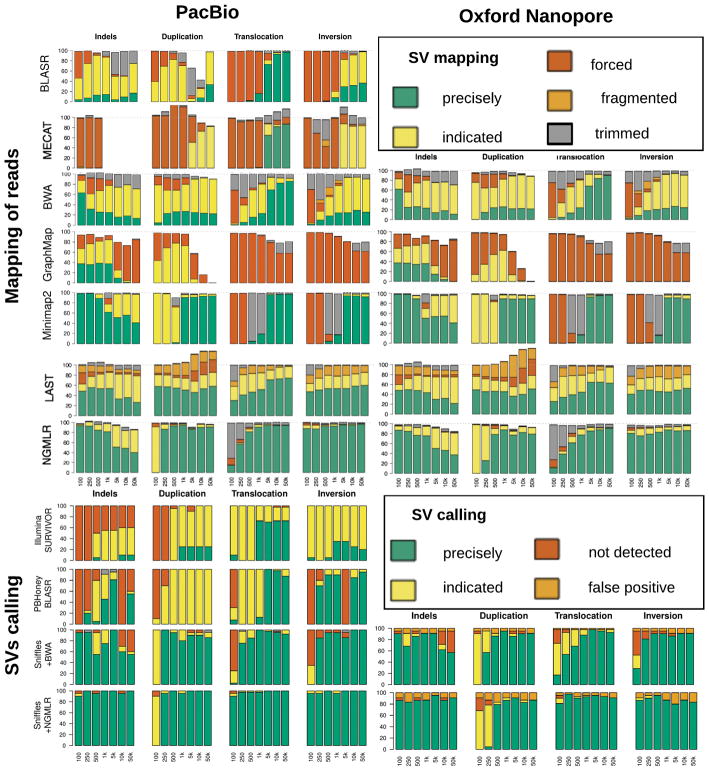
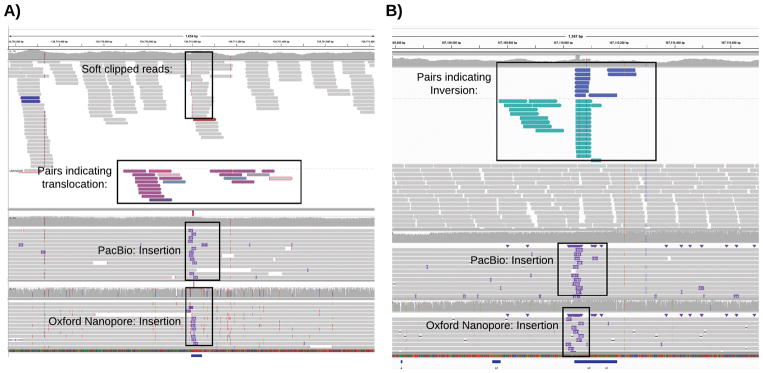
Structural variations are the greatest source of genetic variation, but they remain poorly understood because of technological limitations. Single-molecule long-read sequencing has the potential to dramatically advance the field, although high error rates are a challenge with existing methods. Addressing this need, we introduce open-source methods for long-read alignment (NGMLR; https://github.com/philres/ngmlr ) and structural variant identification (Sniffles; https://github.com/fritzsedlazeck/Sniffles ) that provide unprecedented sensitivity and precision for variant detection, even in repeat-rich regions and for complex nested events that can have substantial effects on human health. In several long-read datasets, including healthy and cancerous human genomes, we discovered thousands of novel variants and categorized systematic errors in short-read approaches. NGMLR and Sniffles can automatically filter false events and operate on low-coverage data, thereby reducing the high costs that have hindered the application of long reads in clinical and research settings.
结构变异是遗传变异的最大来源,但由于技术限制,它们仍然知之甚少。单分子长读测序有可能极大地推动这一领域的发展,尽管现有方法存在高错误率的挑战。为了满足这一需求,我们引入了用于长读对齐(NGMLR;https://github.com/philres/ngmlr)和结构变异识别(Sniffles;https://github.com/fritzsedlazeck/Sniffles)的开源方法,即使在重复丰富的区域和对人类健康有重大影响的复杂嵌套事件中,这些方法也为变异检测提供了前所未有的灵敏度和精度。在包括健康和癌症人类基因组在内的几个长读数据集上,我们发现了数千种新的变异,并对短读方法中的系统错误进行了分类。NGMLR 和 Sniffles 可以自动过滤虚假事件,并在低覆盖数据上运行,从而降低了阻碍长读在临床和研究环境中应用的高成本。