Faculty of Chemistry, University of Wrocław, ul. F. Joliot-Curie 14, 50-383 Wrocław, Poland.
Molecules. 2021 Sep 17;26(18):5642. doi: 10.3390/molecules26185642.
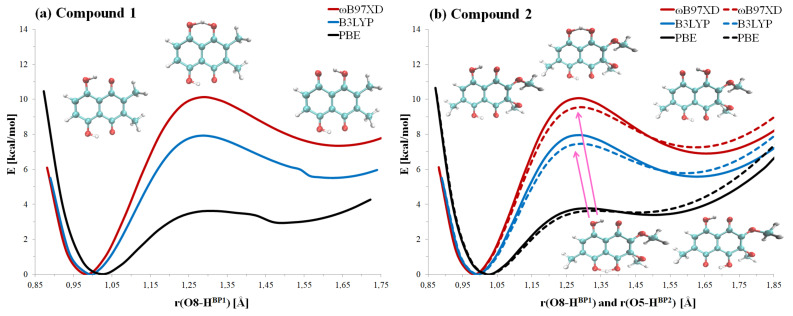
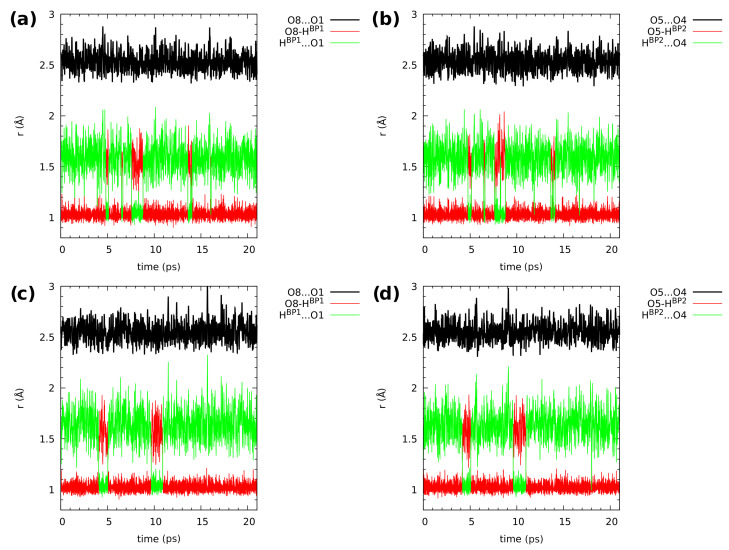
Our long-term investigations have been devoted the characterization of intramolecular hydrogen bonds in cyclic compounds. Our previous work covers naphthazarin, the parent compound of two systems discussed in the current work: 2,3-dimethylnaphthazarin () and 2,3-dimethoxy-6-methylnaphthazarin (). Intramolecular hydrogen bonds and substituent effects in these compounds were analyzed on the basis of Density Functional Theory (DFT), Møller-Plesset second-order perturbation theory (MP2), Coupled Clusters with Singles and Doubles (CCSD) and Car-Parrinello Molecular Dynamics (CPMD). The simulations were carried out in the gas and crystalline phases. The nuclear quantum effects were incorporated a posteriori using the snapshots taken from ab initio trajectories. Further, they were used to solve a vibrational Schrödinger equation. The proton reaction path was studied using B3LYP, ωB97XD and PBE functionals with a 6-311++G(2d,2p) basis set. Two energy minima (deep and shallow) were found, indicating that the proton transfer phenomena could occur in the electronic ground state. Next, the electronic structure and topology were examined in the molecular and proton transferred (PT) forms. The Atoms In Molecules (AIM) theory was employed for this purpose. It was found that the hydrogen bond is stronger in the proton transferred (PT) forms. In order to estimate the dimers' stabilization and forces responsible for it, the Symmetry-Adapted Perturbation Theory (SAPT) was applied. The energy decomposition revealed that dispersion is the primary factor stabilizing the dimeric forms and crystal structure of both compounds. The CPMD results showed that the proton transfer phenomena occurred in both studied compounds, as well as in both phases. In the case of compound , the proton transfer events are more frequent in the solid state, indicating an influence of the environmental effects on the bridged proton dynamics. Finally, the vibrational signatures were computed for both compounds using the CPMD trajectories. The Fourier transformation of the autocorrelation function of atomic velocity was applied to obtain the power spectra. The IR spectra show very broad absorption regions between 700 cm-1-1700 cm-1 and 2300 cm-1-3400 cm-1 in the gas phase and 600 cm-1-1800 cm-1 and 2200 cm-1-3400 cm-1 in the solid state for compound . The absorption regions for compound were found as follows: 700 cm-1-1700 cm-1 and 2300 cm-1-3300 cm-1 for the gas phase and one broad absorption region in the solid state between 700 cm-1 and 3100 cm-1. The obtained spectroscopic features confirmed a strong mobility of the bridged protons. The inclusion of nuclear quantum effects showed a stronger delocalization of the bridged protons.
我们的长期研究致力于对环状化合物中分子内氢键的特征进行研究。我们之前的工作涵盖了萘并恶嗪,这是当前工作中讨论的两个系统的母体化合物:2,3-二甲基萘并恶嗪()和 2,3-二甲氧基-6-甲基萘并恶嗪()。我们基于密度泛函理论(DFT)、Møller-Plesset 二级微扰理论(MP2)、耦合簇与单重和双重(CCSD)和 Car-Parrinello 分子动力学(CPMD),对这些化合物中的分子内氢键和取代基效应进行了分析。模拟在气相和晶相下进行。利用从头算轨迹中的快照,采用后验方法纳入核量子效应。此外,它们还被用于求解振动薛定谔方程。使用 B3LYP、ωB97XD 和 PBE 泛函以及 6-311++G(2d,2p)基组研究了质子反应路径。发现了两个能量最小值(深和浅),表明质子转移现象可能在电子基态中发生。接下来,在分子和质子转移(PT)形式下检查了电子结构和拓扑结构。为此目的采用了分子中的原子(AIM)理论。发现氢键在质子转移(PT)形式中更强。为了估计二聚体的稳定性及其稳定的力,应用了对称适应微扰理论(SAPT)。能量分解表明,色散是稳定二聚体形式和两种化合物晶体结构的主要因素。CPMD 结果表明,质子转移现象发生在两种研究的化合物中,以及在两种相态中。对于化合物,质子转移事件在固态中更为频繁,这表明环境效应对桥接质子动力学的影响。最后,使用 CPMD 轨迹计算了两种化合物的振动特征。应用原子速度自相关函数的傅里叶变换得到功率谱。在气相中,IR 光谱在 700 cm-1-1700 cm-1 和 2300 cm-1-3400 cm-1 之间以及在固态中在 600 cm-1-1800 cm-1 和 2200 cm-1-3400 cm-1 之间显示出非常宽的吸收区域;对于化合物,发现吸收区域如下:气相中的 700 cm-1-1700 cm-1 和 2300 cm-1-3300 cm-1,以及固态中的一个宽吸收区域在 700 cm-1 和 3100 cm-1 之间。所得光谱特征证实了桥接质子的强迁移性。纳入核量子效应显示桥接质子的离域更强。