Faculty of Chemistry, University of Wrocław, ul. F. Joliot-Curie 14, 50-383 Wrocław, Poland.
Faculty of Chemistry, University of Warsaw, ul. Pasteura 1, 01-224 Warsaw, Poland.
Molecules. 2022 Jan 25;27(3):792. doi: 10.3390/molecules27030792.
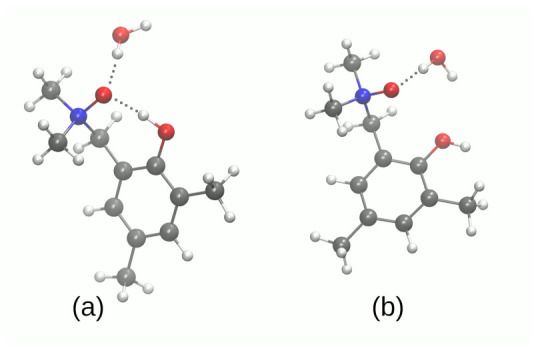
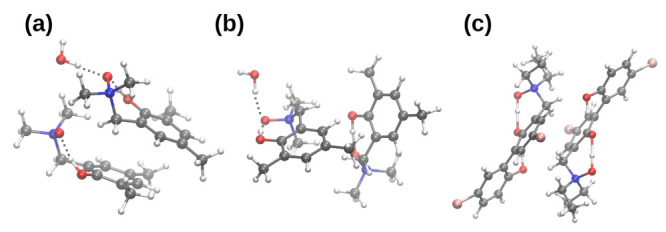
Intra- and intermolecular interactions have been explored in selected -oxide derivatives: 2-(N,N-dimethylamino-N-oxymethyl)-4,6-dimethylphenyl () and 5,5'-dibromo-3-diethylaminomethyl-2,2'-biphenol -oxide (). Both compounds possess intramolecular hydrogen bonding, which is classified as moderate in and strong in , and resonance-assisted in both cases. Density Functional Theory (DFT) in its classical formulation as well as Time-Dependent extension (TD-DFT) were employed to study proton transfer phenomena. The simulations were performed in the gas phase and with implicit and explicit solvation models. The obtained structures of the studied -oxides were compared with experimental data available. The proton reaction path was investigated using scan with an optimization method, and water molecule reorientation in the monohydrate of was found upon the proton scan progress. It was found that spontaneous proton transfer phenomenon cannot occur in the electronic ground state of the compound . An opposite situation was noticed for the compound . The changes of nucleophilicity and electrophilicity upon the bridged proton migration were analyzed on the basis of Fukui functions in the case of . The interaction energy decomposition of dimers and microsolvation models was investigated using Symmetry-Adapted Perturbation Theory (SAPT). The simulations were performed in both phases to introduce polar environment influence on the interaction energies. The SAPT study showed rather minor role of induction in the formation of homodimers. However, it is worth noticing that the same induction term is responsible for the preference of water molecules' interaction with -oxide hydrogen bond acceptor atoms in the microsolvation study. The Natural Bond Orbital (NBO) analysis was performed for the complexes with water to investigate the charge flow upon the polar environment introduction. Finally, the TD-DFT was applied for isolated molecules as well as for microsolvation models showing that the presence of solvent affects excited states, especially when the -oxide acceptor atom is microsolvated.
已在选定的 -氧化物衍生物中探索了分子内和分子间相互作用:2-(N,N-二甲基氨基-N-羟甲基)-4,6-二甲基苯基()和 5,5'-二溴-3-二乙氨基甲基-2,2'-联苯酚 -氧化物()。这两种化合物都具有分子内氢键,在 中为中等强度,在 中为强氢键,在两种情况下均为共振辅助。采用经典形式的密度泛函理论(DFT)及其时间相关扩展(TD-DFT)来研究质子转移现象。模拟在气相中进行,并采用隐式和显式溶剂化模型。将研究的 -氧化物的获得结构与可用的实验数据进行比较。使用扫描法研究质子反应路径,并使用优化方法,发现质子扫描过程中一水合物中的水分子重新取向。发现质子转移现象不能在化合物的电子基态中自发发生。对于化合物,则出现相反的情况。基于福井函数分析了桥接质子迁移时亲核性和电负性的变化。使用对称自适应微扰理论(SAPT)研究了二聚体的相互作用能分解和微溶剂化模型。在两种相中进行模拟,以引入极性环境对相互作用能的影响。SAPT 研究表明,诱导在同二聚体的形成中仅起次要作用。但是,值得注意的是,在微溶剂化研究中,相同的诱导项负责水分子与 -氧化物氢键接受原子相互作用的偏好。进行自然键轨道(NBO)分析,以研究引入极性环境时电荷的流动。最后,将 TD-DFT 应用于孤立分子和微溶剂化模型,结果表明,溶剂的存在会影响激发态,特别是当 -氧化物接受体原子被微溶剂化时。