Wang Min, Zhu Jie, Zhao Fang, Xiao Jiani
Department of Respiratory and Geriatrics, Chongqing Public Health Medical Center, Chongqing, China.
Department of Respiratory and Critical Care Medicine, West China Hospital, Sichuan University, Chengdu, China.
Front Oncol. 2021 Sep 14;11:643503. doi: 10.3389/fonc.2021.643503. eCollection 2021.
With the development and application of targeted therapies like tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs), non-small cell lung cancer (NSCLC) patients have achieved remarkable survival benefits in recent years. However, epidermal growth factor receptor (EGFR) wild-type and low expression of programmed death-ligand 1 (PD-L1) NSCLCs remain unmanageable. Few treatments for these patients exist, and more side effects with combination therapies have been observed. We intended to generate a metabolic gene signature that could successfully identify high-risk patients and reveal its underlying molecular immunology characteristics.
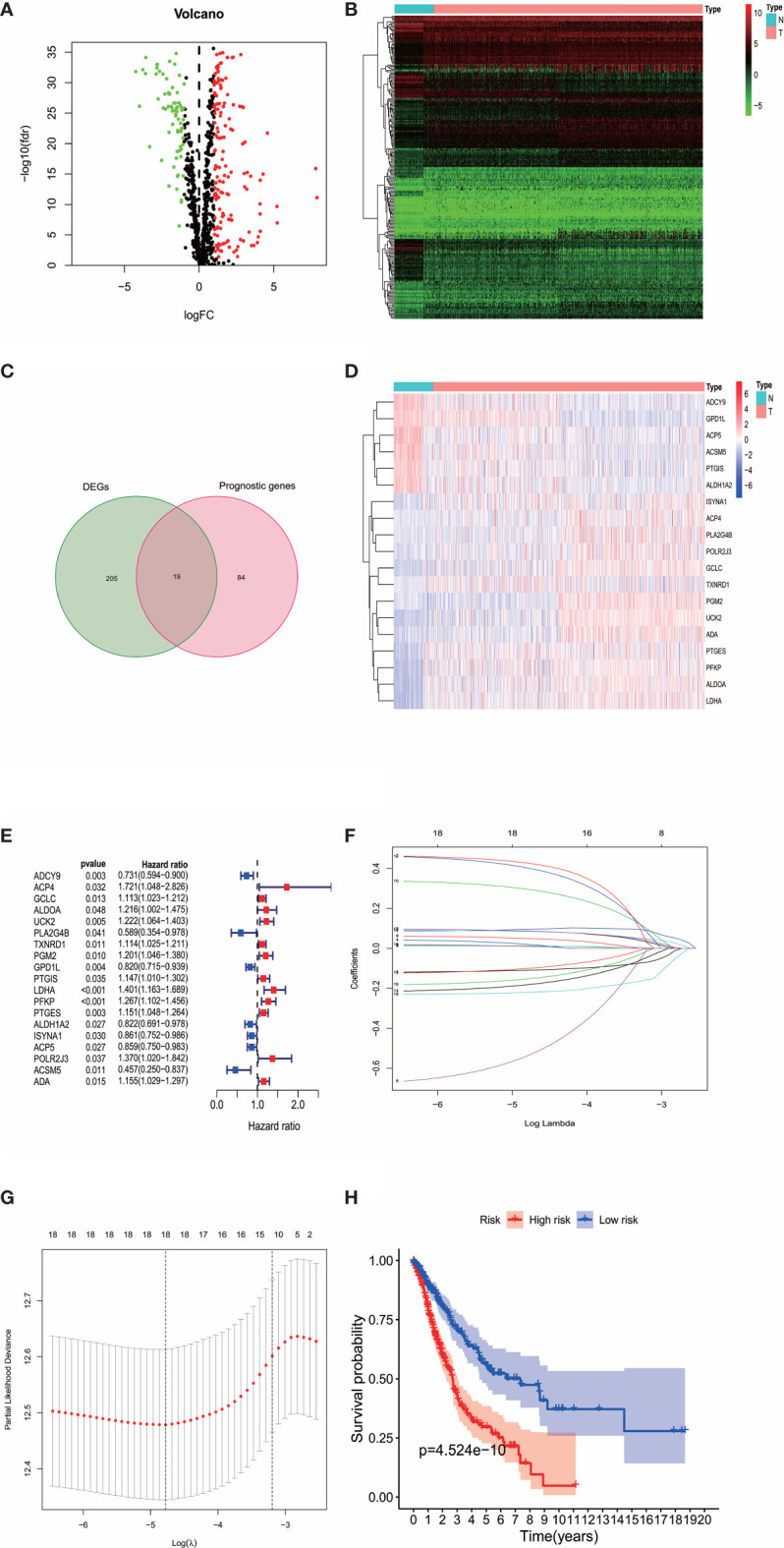
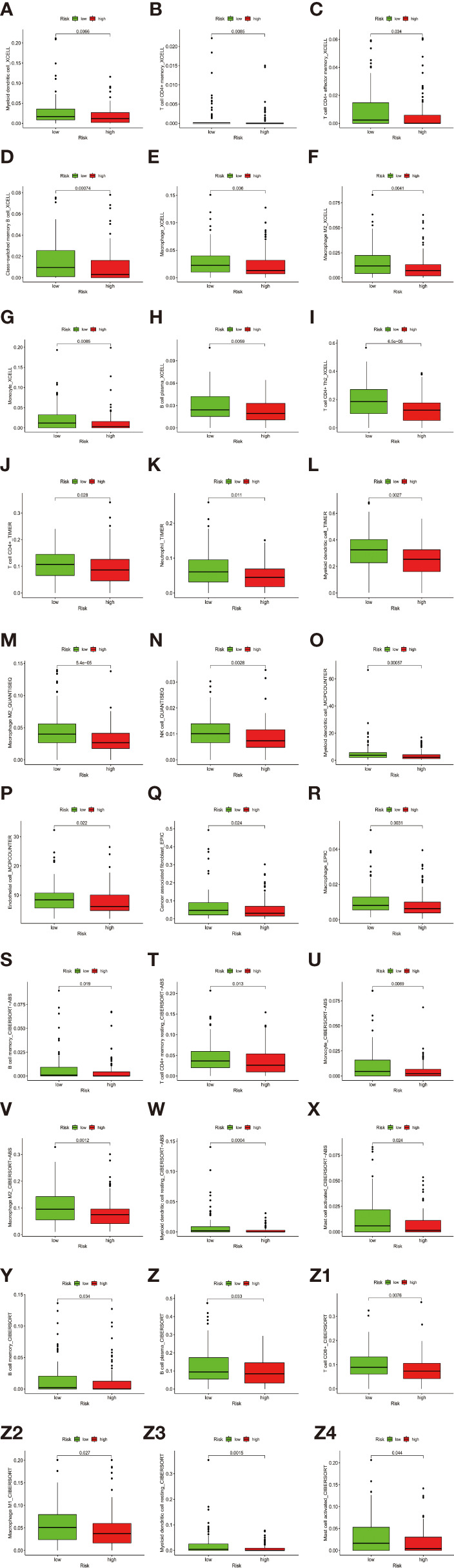
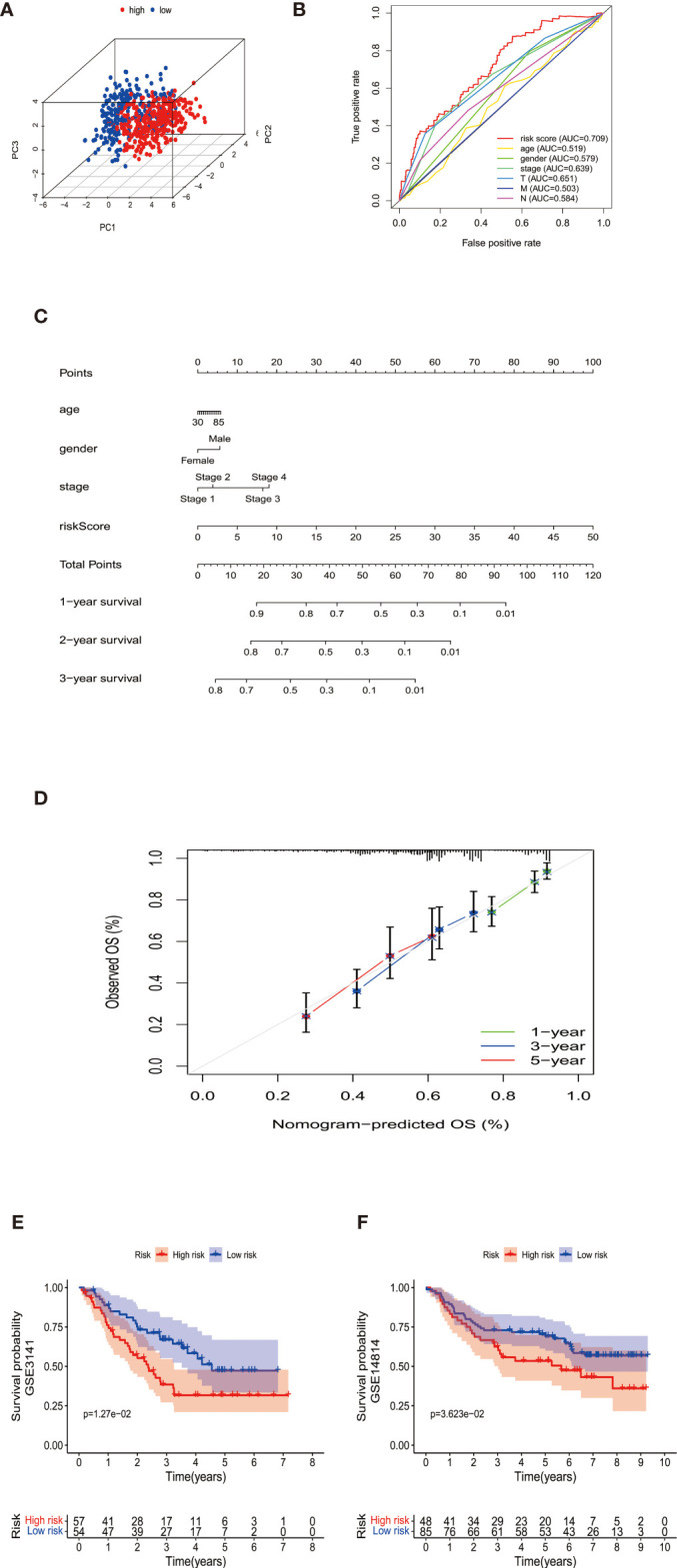
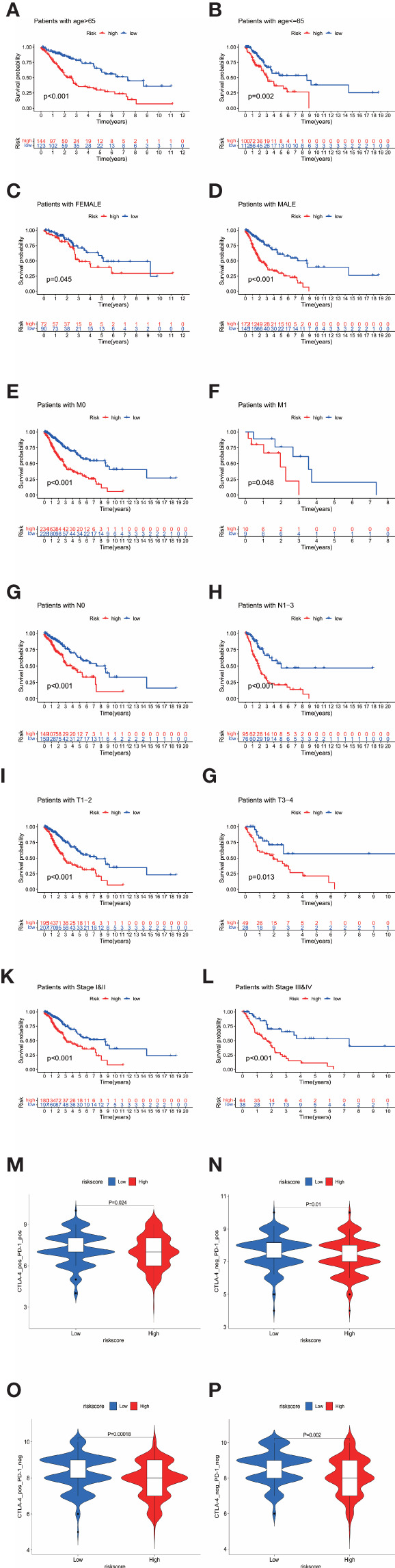
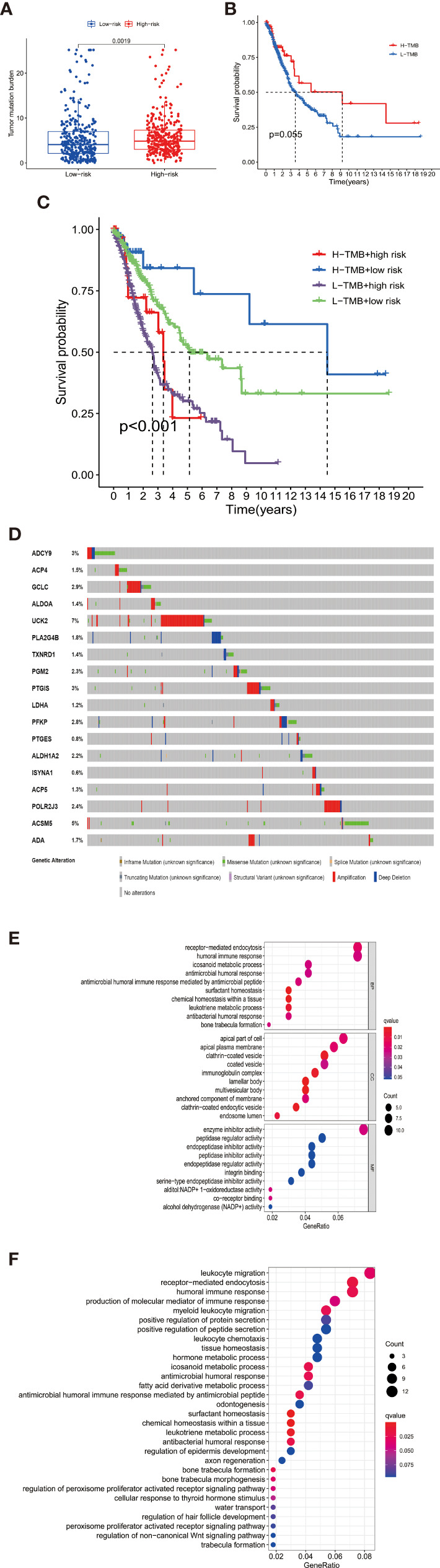
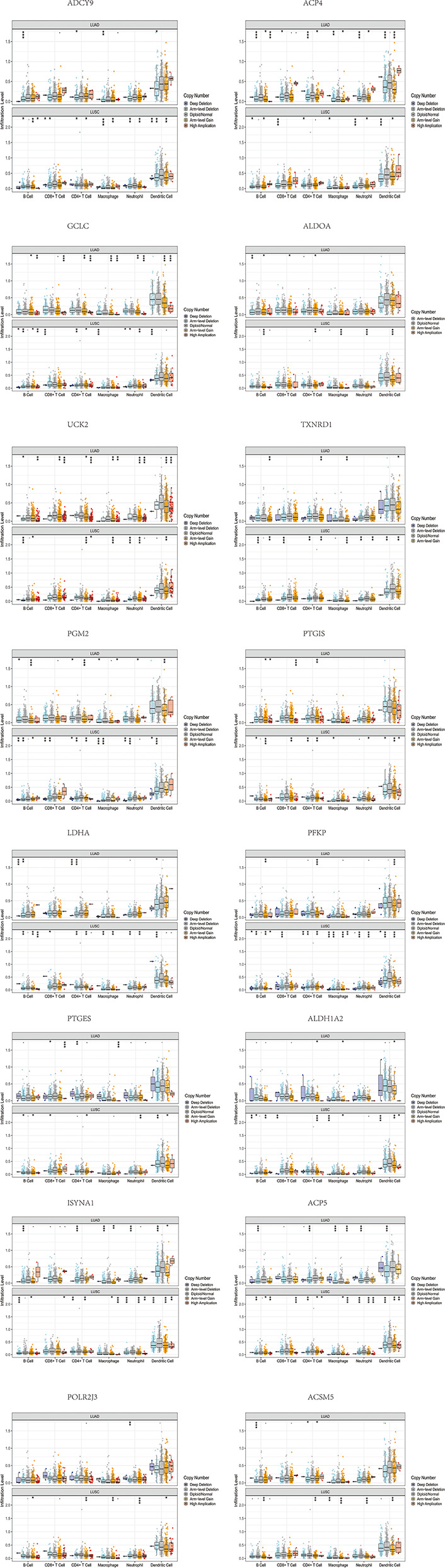
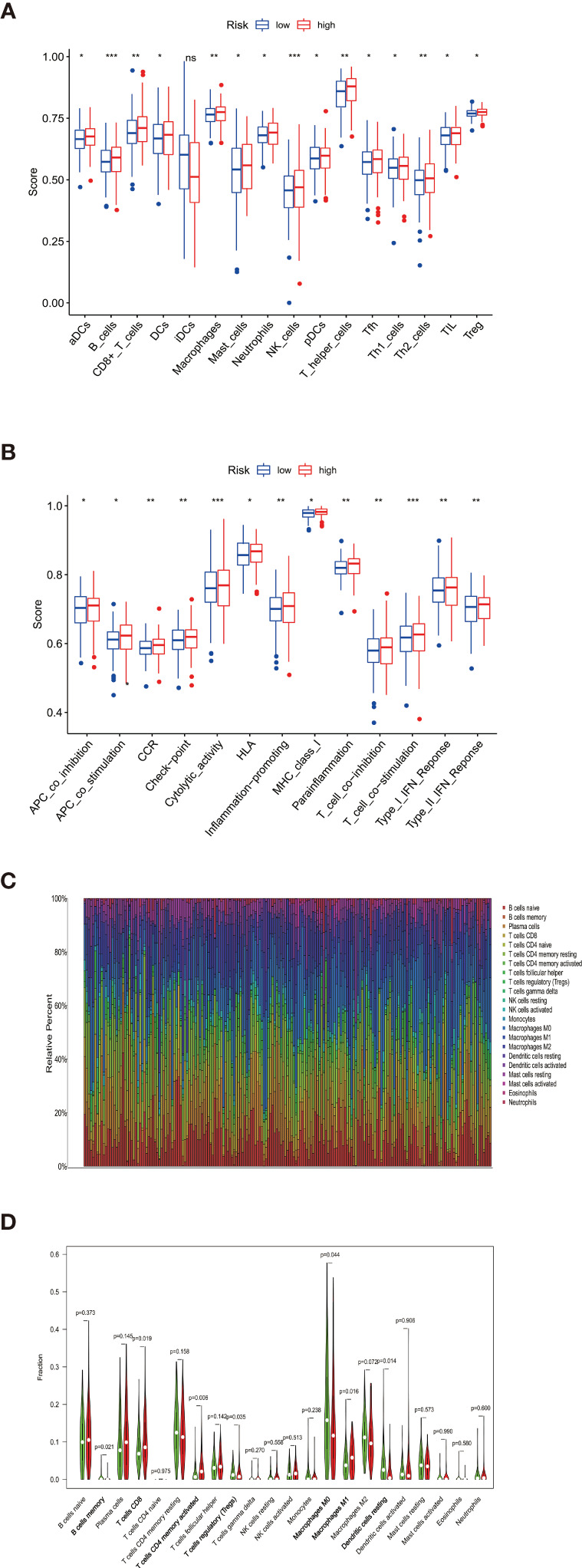
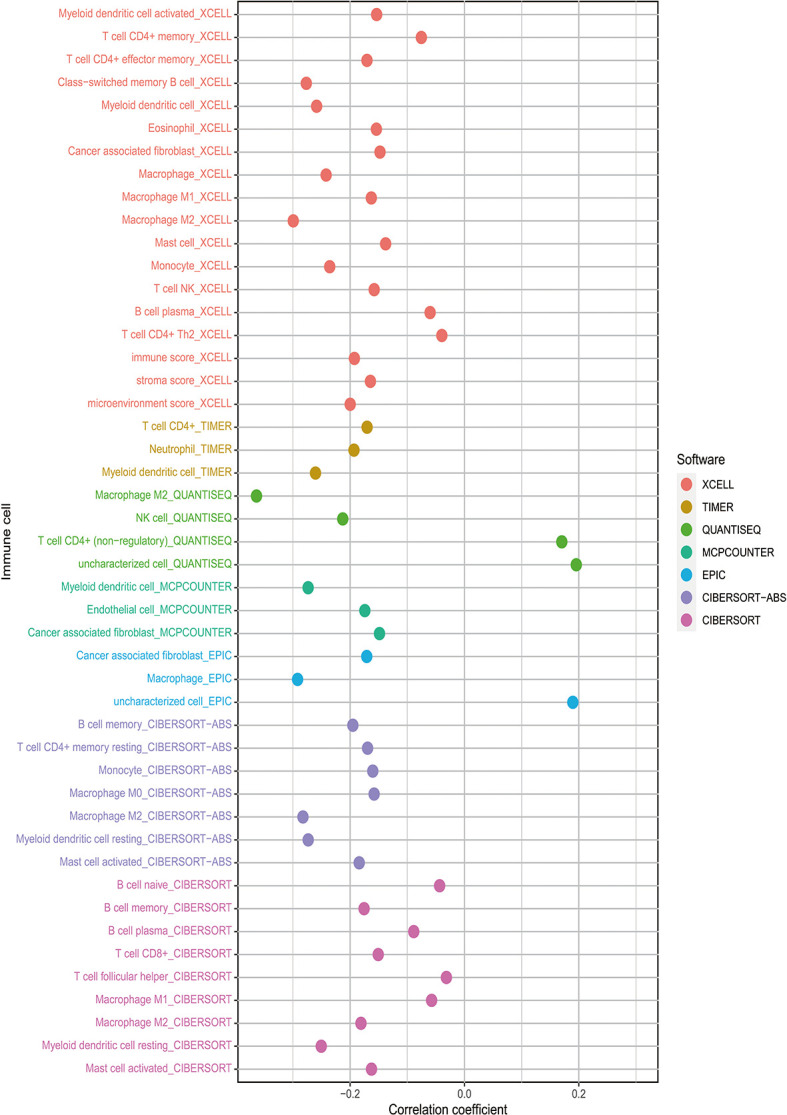
By identifying the bottom 50% PD-L1 expression level as PD-L1 low expression and removing EGFR mutant samples, a total of 640 lung adenocarcinoma (LUAD) and lung squamous carcinoma (LUSC) tumor samples and 93 adjacent non-tumor samples were finally extracted from The Cancer Genome Atlas (TCGA). We identified differentially expressed metabolic genes (DEMGs) by R package limma and the prognostic genes by Univariate Cox proportional hazards regression analyses. The intersect genes between DEMGs and prognostic genes were put into the least absolute shrinkage and selection operator (LASSO) penalty Cox regression analysis. The metabolic gene signature contained 18 metabolic genes generated and successfully stratified LUAD and LUSC patients into the high-risk and low-risk groups, which was also validated by the Gene Expression Omnibus (GEO) database. Its accuracy was proved by the time-dependent Receiver Operating Characteristic (ROC) curve, Principal Components Analysis (PCA), and nomogram. Furthermore, the Single-sample Gene Set Enrichment Analysis (ssGSEA) and diverse acknowledged methods include XCELL, TIMER, QUANTISEQ, MCPcounter, EPIC, CIBERSORT-ABS, and CIBERSORT revealed its underlying antitumor immunosuppressive status. Besides, its relationship with somatic copy number alterations (SCNAs) and tumor mutational burden (TMB) was also discussed.
It is noteworthy that metabolism reprogramming is associated with the survival of the double-negative LUAD and LUSC patients. The SCNAs and TMB of critical metabolic genes can inhibit the antitumor immune process, which might be a promising therapeutic target.
随着酪氨酸激酶抑制剂(TKIs)和免疫检查点抑制剂(ICIs)等靶向治疗的发展与应用,近年来非小细胞肺癌(NSCLC)患者获得了显著的生存益处。然而,表皮生长因子受体(EGFR)野生型和程序性死亡配体1(PD-L1)低表达的NSCLC患者仍然难以治疗。针对这些患者的治疗方法很少,并且联合治疗观察到更多的副作用。我们旨在生成一种代谢基因特征,该特征可以成功识别高危患者并揭示其潜在的分子免疫学特征。
通过将PD-L1表达水平最低的50%定义为PD-L1低表达,并去除EGFR突变样本,最终从癌症基因组图谱(TCGA)中提取了总共640例肺腺癌(LUAD)和肺鳞状细胞癌(LUSC)肿瘤样本以及93例相邻的非肿瘤样本。我们通过R包limma鉴定差异表达的代谢基因(DEMGs),并通过单变量Cox比例风险回归分析鉴定预后基因。将DEMGs和预后基因之间的交集基因进行最小绝对收缩和选择算子(LASSO)惩罚Cox回归分析。代谢基因特征包含18个代谢基因,这些基因成功地将LUAD和LUSC患者分为高危组和低危组,这也通过基因表达综合数据库(GEO)进行了验证。其准确性通过时间依赖性受试者工作特征(ROC)曲线、主成分分析(PCA)和列线图得到证明。此外,单样本基因集富集分析(ssGSEA)以及多种公认的方法,包括XCELL、TIMER、QUANTISEQ、MCPcounter、EPIC、CIBERSORT-ABS和CIBERSORT,揭示了其潜在的抗肿瘤免疫抑制状态。此外,还讨论了其与体细胞拷贝数改变(SCNAs)和肿瘤突变负担(TMB)的关系。
值得注意的是,代谢重编程与双阴性LUAD和LUSC患者的生存相关。关键代谢基因的SCNAs和TMB可抑制抗肿瘤免疫过程,这可能是一个有前景的治疗靶点。