Xu Ran, Kang Le, Wei Siang, Yang Chunjie, Fu Yuanfeng, Ding Zhiwen, Zou Yunzeng
Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, Shanghai, China.
Front Cardiovasc Med. 2021 Sep 22;8:748156. doi: 10.3389/fcvm.2021.748156. eCollection 2021.
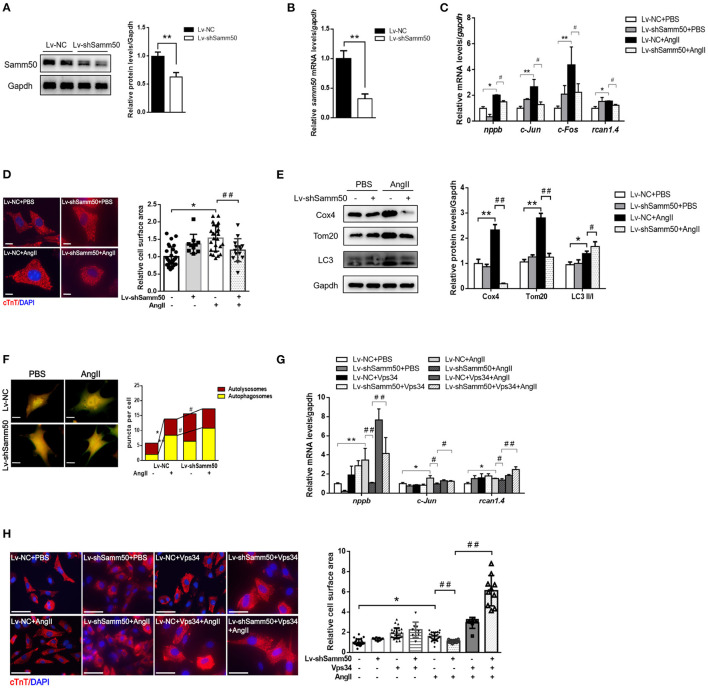
Pathological cardiac hypertrophy, the adaptive response of the myocardium to various pathological stimuli, is one of the primary predictors and predisposing factors of heart failure. However, its molecular mechanisms underlying pathogenesis remain poorly understood. Here, we studied the function of Samm50 in mitophagy during Ang II-induced cardiomyocyte hypertrophy via lentiviruses mediated knockdown and overexpression of Samm50 protein. We first found that Samm50 is a key positive regulator of cardiac hypertrophy, for western blot and real-time quantitative PCR detection revealed Samm50 was downregulated both in pressure-overload-induced hypertrophic hearts and Ang II-induced cardiomyocyte hypertrophy. Then, Samm50 overexpression exhibits enhanced induction of cardiac hypertrophy marker genes and cell enlargement in primary mouse cardiomyocytes by qPCR and immunofluorescence analysis, respectively. Meanwhile, Samm50 remarkably reduced Ang II-induced autophagy as indicated by decreased mitophagy protein levels and autophagic flux, whereas the opposite phenotype was observed in Samm50 knockdown cardiomyocytes. However, the protective role of Samm50 deficiency against cardiac hypertrophy was abolished by inhibiting mitophagy through Vps34 inhibitor or Pink1 knockdown. Moreover, we further demonstrated that Samm50 interacted with Pink1 and stimulated the accumulation of Parkin on mitochondria to initiate mitophagy by co-immunoprecipitation analysis and immunofluorescence. Thus, these results suggest that Samm50 regulates Pink1-Parkin-mediated mitophagy to promote cardiac hypertrophy, and targeting mitophagy may provide new insights into the treatment of cardiac hypertrophy.
病理性心脏肥大是心肌对各种病理刺激的适应性反应,是心力衰竭的主要预测指标和易感因素之一。然而,其发病机制的分子机制仍知之甚少。在此,我们通过慢病毒介导的Samm50蛋白敲低和过表达,研究了Samm50在血管紧张素II诱导的心肌细胞肥大过程中对线粒体自噬的作用。我们首先发现Samm50是心脏肥大的关键正向调节因子,因为蛋白质印迹和实时定量PCR检测显示,在压力超负荷诱导的肥大心脏和血管紧张素II诱导的心肌细胞肥大中,Samm50均下调。然后,分别通过qPCR和免疫荧光分析,Samm50过表达在原代小鼠心肌细胞中表现出增强的心脏肥大标记基因诱导和细胞增大。同时,线粒体自噬蛋白水平和自噬通量降低表明,Samm50显著减少了血管紧张素II诱导的自噬,而在Samm50敲低的心肌细胞中观察到相反的表型。然而,通过Vps34抑制剂或Pink1敲低抑制线粒体自噬,消除了Samm50缺陷对心脏肥大的保护作用。此外,我们通过免疫共沉淀分析和免疫荧光进一步证明,Samm50与Pink1相互作用,并刺激Parkin在线粒体上的积累以启动线粒体自噬。因此,这些结果表明,Samm50调节Pink1-Parkin介导的线粒体自噬以促进心脏肥大,靶向线粒体自噬可能为心脏肥大的治疗提供新的见解。