Faculty of Bioengineering, Lomonosov Moscow State University, 119234 Moscow, Russia.
Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia.
Molecules. 2021 Sep 26;26(19):5839. doi: 10.3390/molecules26195839.
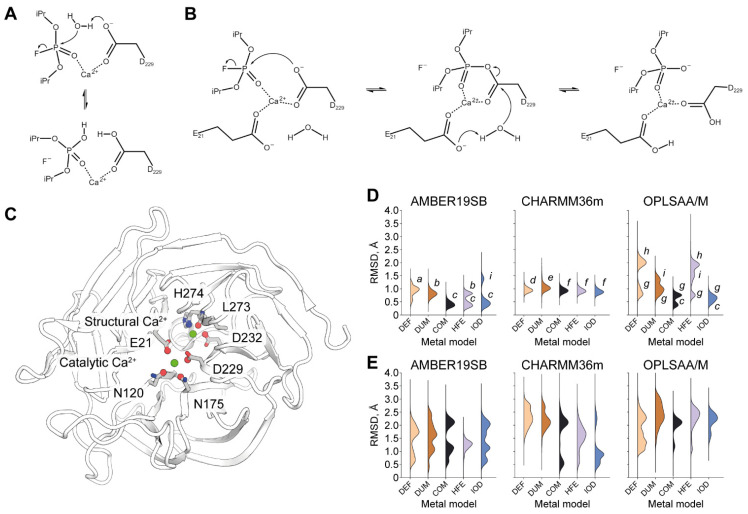
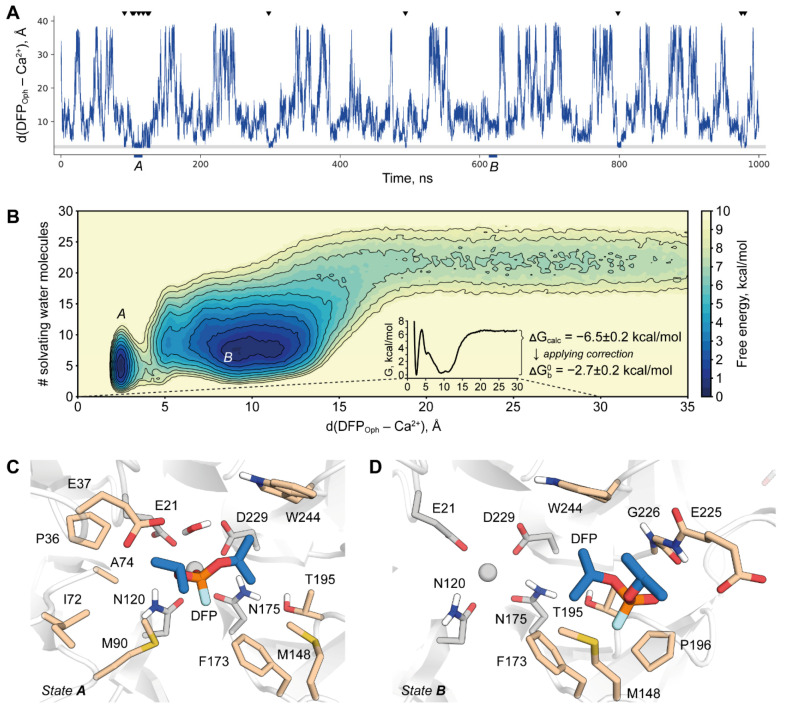
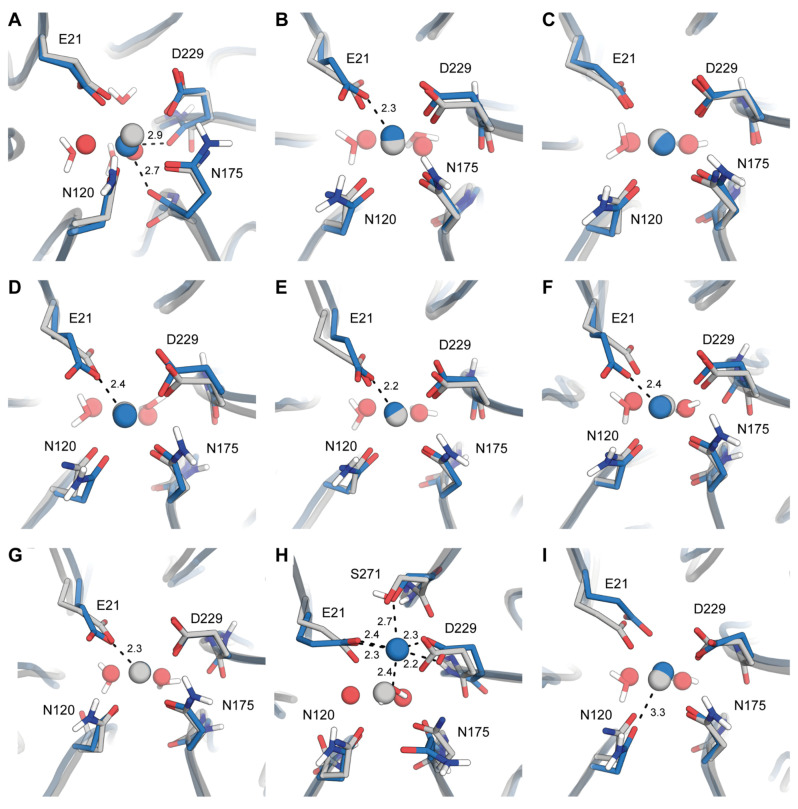
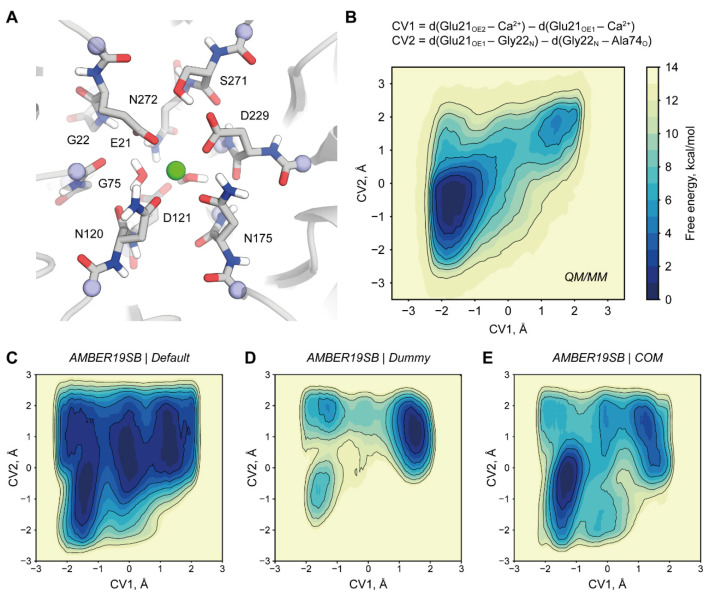
Organophosphate hydrolases are promising as potential biotherapeutic agents to treat poisoning with pesticides or nerve gases. However, these enzymes often need to be further engineered in order to become useful in practice. One example of such enhancement is the alteration of enantioselectivity of diisopropyl fluorophosphatase (DFPase). Molecular modeling techniques offer a unique opportunity to address this task rationally by providing a physical description of the substrate-binding process. However, DFPase is a metalloenzyme, and correct modeling of metal cations is a challenging task generally coming with a tradeoff between simulation speed and accuracy. Here, we probe several molecular mechanical parameter combinations for their ability to empower long simulations needed to achieve a quantitative description of substrate binding. We demonstrate that a combination of the Amber19sb force field with the recently developed 12-6 Ca models allows us to both correctly model DFPase and obtain new insights into the DFP binding process.
有机磷水解酶有望成为治疗农药或神经毒气中毒的潜在生物治疗剂。然而,为了在实践中实际应用,这些酶通常需要进一步进行工程化改造。改变二异丙基氟磷酸酶(DFPase)的对映选择性就是这样一种增强的例子。分子建模技术通过提供对底物结合过程的物理描述,为合理解决这一任务提供了独特的机会。然而,DFPase 是一种金属酶,正确模拟金属阳离子通常是一项具有挑战性的任务,通常需要在模拟速度和准确性之间进行权衡。在这里,我们研究了几种分子力学参数组合,以确定它们在实现对底物结合的定量描述所需的长时间模拟中的能力。我们证明,Amber19sb 力场与最近开发的 12-6Ca 模型的结合,使我们既能正确地模拟 DFPase,又能深入了解 DFP 的结合过程。