Biology Centre of the Czech Academy of Sciences, Institute of Entomology, České Budějovice, Czech Republic.
Faculty of Science, University of South Bohemia, České Budějovice, Czech Republic.
Mol Ecol. 2021 Dec;30(23):6021-6035. doi: 10.1111/mec.16240. Epub 2021 Oct 31.
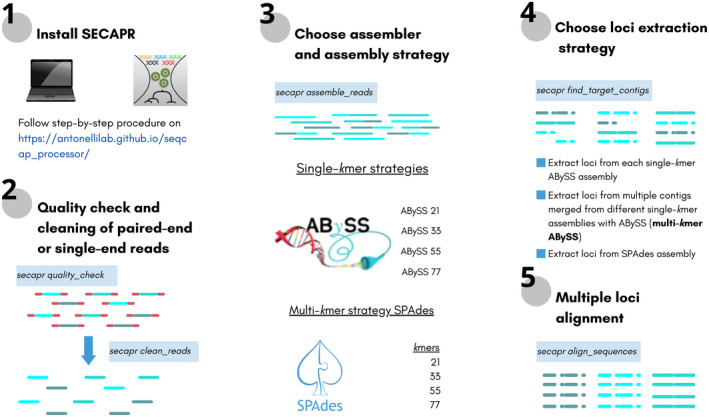
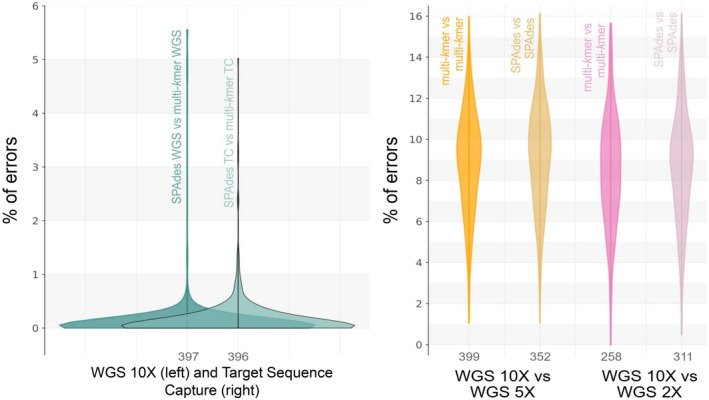
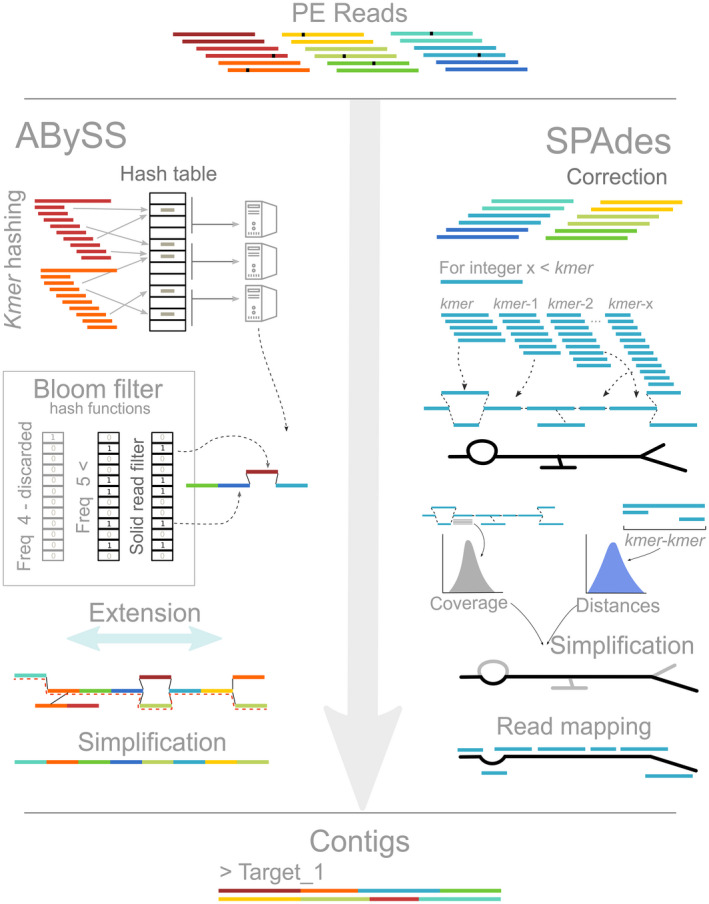
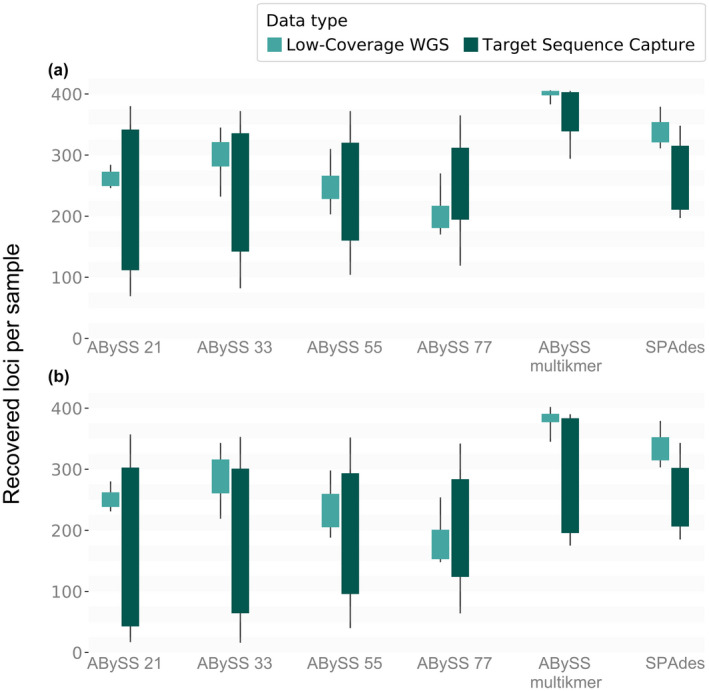
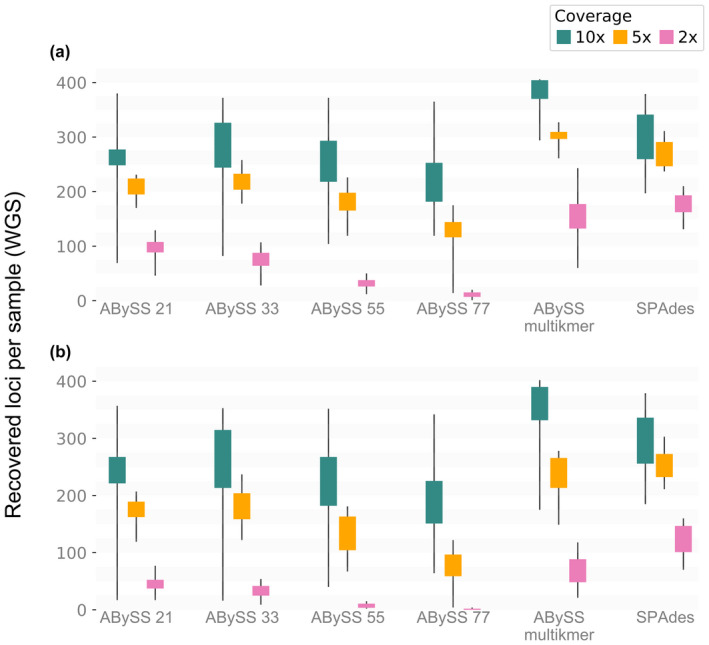
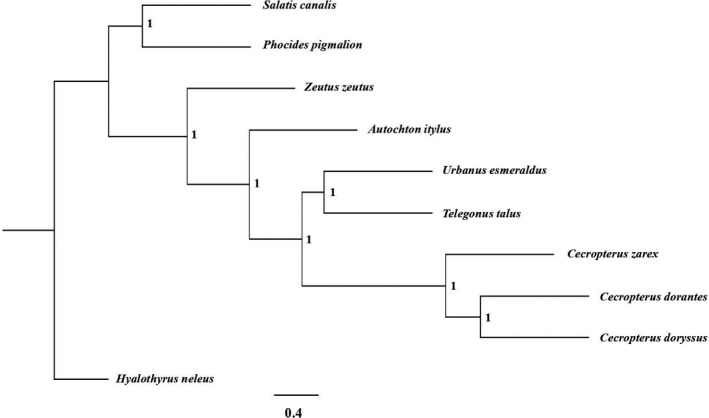
The increasing availability of short-read whole genome sequencing (WGS) provides unprecedented opportunities to study ecological and evolutionary processes. Although loci of interest can be extracted from WGS data and combined with target sequence data, this requires suitable bioinformatic workflows. Here, we test different assembly and locus extraction strategies and implement them into secapr, a pipeline that processes short-read data into multilocus alignments for phylogenetics and molecular ecology analyses. We integrate the processing of data from low-coverage WGS (<30×) and target sequence capture into a flexible framework, while optimizing de novo contig assembly and loci extraction. Specifically, we test different assembly strategies by contrasting their ability to recover loci from targeted butterfly protein-coding genes, using four data sets: a WGS data set across different average coverages (10×, 5× and 2×) and a data set for which these loci were enriched prior to sequencing via target sequence capture. Using the resulting de novo contigs, we account for potential errors within contigs and infer phylogenetic trees to evaluate the ability of each assembly strategy to recover species relationships. We demonstrate that choosing multiple sizes of kmer simultaneously for assembly results in the highest yield of extracted loci from de novo assembled contigs, while data sets derived from sequencing read depths as low as 5× recovers the expected species relationships in phylogenetic trees. By making the tested assembly approaches available in the secapr pipeline, we hope to inspire future studies to incorporate complementary data and make an informed choice on the optimal assembly strategy.
短读全基因组测序 (WGS) 的日益普及为研究生态和进化过程提供了前所未有的机会。虽然可以从 WGS 数据中提取感兴趣的基因座,并将其与目标序列数据相结合,但这需要合适的生物信息学工作流程。在这里,我们测试了不同的组装和基因座提取策略,并将其实现到 secapr 中,这是一个将短读数据处理成用于系统发育和分子生态学分析的多位点对齐的管道。我们将低覆盖率 WGS(<30×)和目标序列捕获的数据处理集成到一个灵活的框架中,同时优化从头组装和基因座提取。具体来说,我们通过对比它们从目标蝴蝶蛋白编码基因中提取基因座的能力来测试不同的组装策略,使用了四个数据集:一个跨越不同平均覆盖率 (10×、5×和 2×) 的 WGS 数据集,以及一个在测序前通过目标序列捕获富集这些基因座的数据集。使用生成的从头组装的 contigs,我们考虑了 contigs 内部的潜在错误,并推断了系统发育树,以评估每种组装策略恢复物种关系的能力。我们证明,同时选择多个 kmer 大小用于组装可以从从头组装的 contigs 中获得最高的提取基因座产量,而源自测序读深低至 5×的数据集可以在系统发育树中恢复预期的物种关系。通过在 secapr 管道中提供经过测试的组装方法,我们希望激发未来的研究,纳入补充数据,并在最佳组装策略上做出明智的选择。