Department of Biological and Environmental Sciences, University of Gothenburg, SE-413 19 Göteborg, Sweden.
Gothenburg Global Biodiversity Centre, Box 461, SE-405 30 Göteborg, Sweden.
Syst Biol. 2019 Jan 1;68(1):32-46. doi: 10.1093/sysbio/syy039.
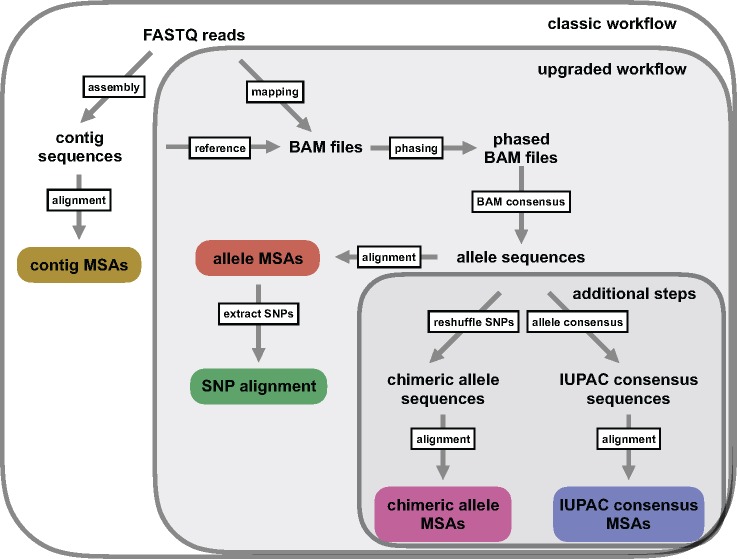
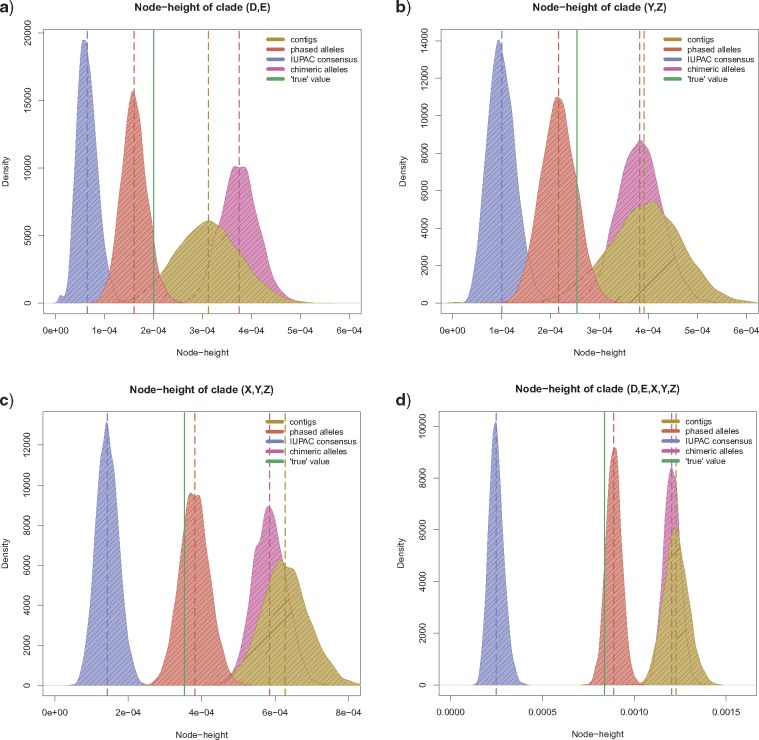
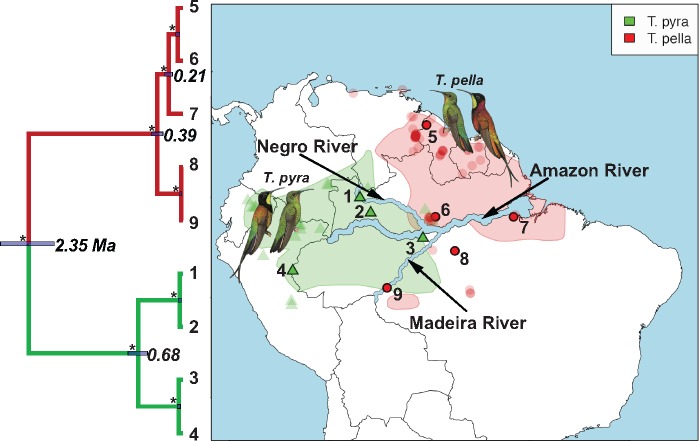
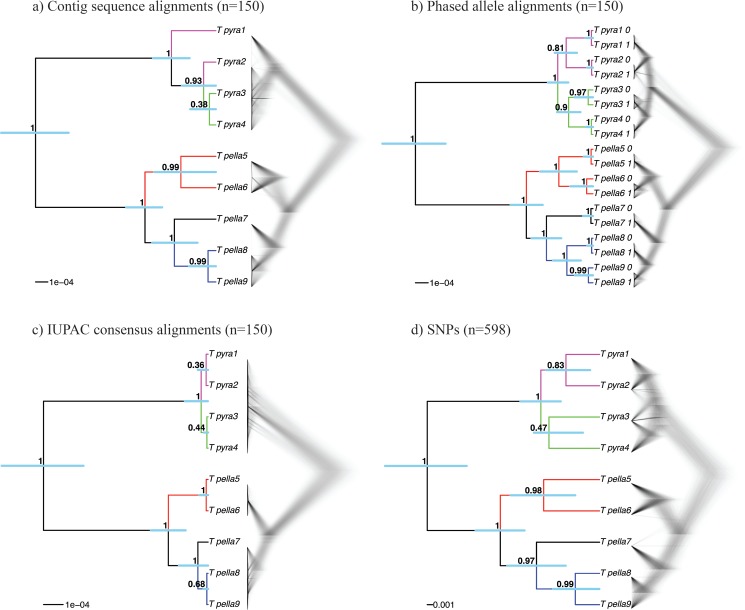
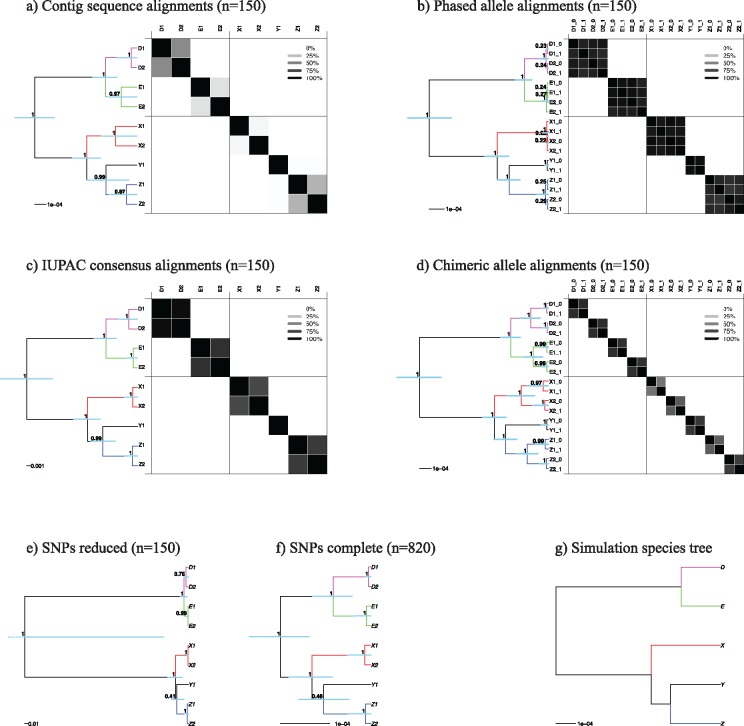
Advances in high-throughput sequencing techniques now allow relatively easy and affordable sequencing of large portions of the genome, even for nonmodel organisms. Many phylogenetic studies reduce costs by focusing their sequencing efforts on a selected set of targeted loci, commonly enriched using sequence capture. The advantage of this approach is that it recovers a consistent set of loci, each with high sequencing depth, which leads to more confidence in the assembly of target sequences. High sequencing depth can also be used to identify phylogenetically informative allelic variation within sequenced individuals, but allele sequences are infrequently assembled in phylogenetic studies. Instead, many scientists perform their phylogenetic analyses using contig sequences which result from the de novo assembly of sequencing reads into contigs containing only canonical nucleobases, and this may reduce both statistical power and phylogenetic accuracy. Here, we develop an easy-to-use pipeline to recover allele sequences from sequence capture data, and we use simulated and empirical data to demonstrate the utility of integrating these allele sequences to analyses performed under the multispecies coalescent model. Our empirical analyses of ultraconserved element locus data collected from the South American hummingbird genus Topaza demonstrate that phased allele sequences carry sufficient phylogenetic information to infer the genetic structure, lineage divergence, and biogeographic history of a genus that diversified during the last 3 myr. The phylogenetic results support the recognition of two species and suggest a high rate of gene flow across large distances of rainforest habitats but rare admixture across the Amazon River. Our simulations provide evidence that analyzing allele sequences leads to more accurate estimates of tree topology and divergence times than the more common approach of using contig sequences.
高通量测序技术的进步使得即使对于非模式生物,也可以相对轻松且经济地对基因组的大部分区域进行测序。许多系统发育研究通过将测序工作集中在一组选定的靶向基因座上来降低成本,这些基因座通常使用序列捕获来富集。这种方法的优点是它可以恢复一组一致的基因座,每个基因座都具有较高的测序深度,从而提高了目标序列组装的可信度。高测序深度还可用于识别测序个体中具有系统发育意义的等位基因变异,但在系统发育研究中,等位基因序列很少被组装。相反,许多科学家使用从头组装测序reads 成仅包含标准核碱基的 contigs 而产生的 contig 序列来进行系统发育分析,这可能会降低统计效力和系统发育准确性。在这里,我们开发了一种易于使用的管道,用于从序列捕获数据中恢复等位基因序列,并使用模拟和实验数据来证明整合这些等位基因序列到多物种合并模型下进行的分析中的效用。我们对从南美蜂鸟属 Topaza 收集的超保守元件基因座数据进行的实证分析表明,相分的等位基因序列携带足够的系统发育信息,可推断一个在过去 300 万年中多样化的属的遗传结构、谱系分化和生物地理历史。系统发育结果支持识别两个物种的观点,并表明在热带雨林栖息地的大范围内存在较高的基因流动,但在亚马逊河流域的基因混合则很少。我们的模拟结果提供了证据,表明分析等位基因序列比使用更常见的 contig 序列的方法更能准确估计树拓扑结构和分歧时间。