Neurogenetics Research Centre, IRCCS Mondino Foundation, Pavia, Italy.
Department of Molecular Medicine, University of Pavia, Pavia, Lombardia, Italy.
J Med Genet. 2022 Sep;59(9):888-894. doi: 10.1136/jmedgenet-2021-108114. Epub 2021 Oct 21.
Joubert syndrome (JS) is a recessively inherited ciliopathy characterised by congenital ocular motor apraxia (COMA), developmental delay (DD), intellectual disability, ataxia, multiorgan involvement, and a unique cerebellar and brainstem malformation. Over 40 JS-associated genes are known with a diagnostic yield of 60%-75%.In 2018, we reported homozygous hypomorphic missense variants of the gene in two families with mild JS. Recently, heterozygous truncating variants were identified in families with dominantly inherited COMA, occasionally associated with mild DD and subtle cerebellar anomalies.
We reanalysed next generation sequencing (NGS) data in two cohorts comprising 1097 probands referred for genetic testing of JS genes.
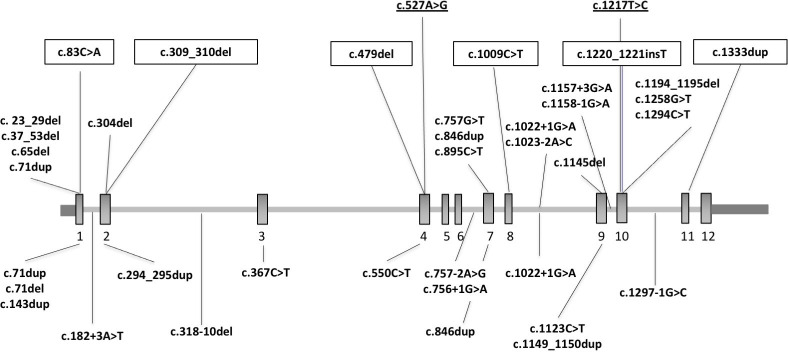
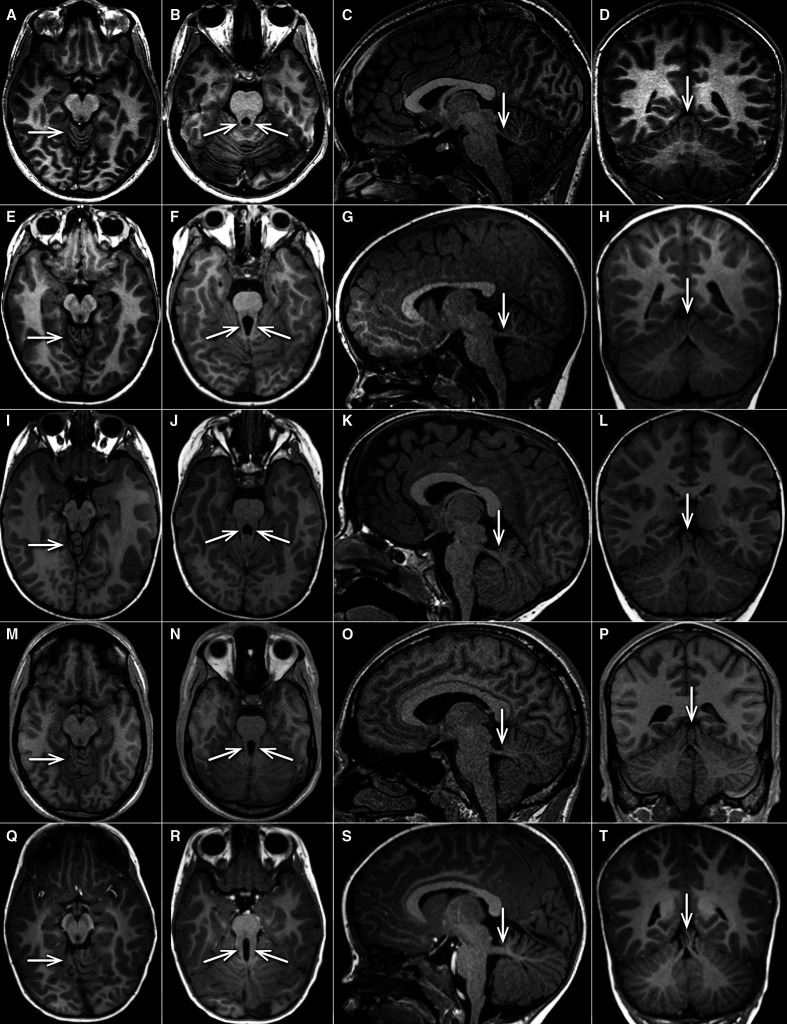
Heterozygous truncating and splice-site variants were detected in 22 patients from 17 families (1.5%) with strong male prevalence (86%), and in 8 asymptomatic parents. Patients presented with COMA, hypotonia, ataxia and mild DD, and only a third manifested intellectual disability of variable severity. Brain MRI showed consistent findings characterised by vermis hypoplasia, superior cerebellar dysplasia and subtle-to-mild abnormalities of the superior cerebellar peduncles. The same pattern was observed in two out of three tested asymptomatic parents.
Heterozygous truncating or splice-site variants cause a novel neurodevelopmental syndrome encompassing COMA and mild JS, which likely represent overlapping entities. Variants can arise de novo or be inherited from a healthy parent, representing the first cause of JS with dominant inheritance and reduced penetrance. Awareness of this condition will increase the diagnostic yield of JS genetic testing, and allow appropriate counselling about prognosis, medical monitoring and recurrence risk.
杰特综合征(JS)是一种常染色体隐性遗传的纤毛病,其特征为先天性眼球运动失调(COMA)、发育迟缓(DD)、智力障碍、共济失调、多器官受累以及独特的小脑和脑干畸形。已知有超过 40 种与 JS 相关的基因,其诊断率为 60%-75%。2018 年,我们报道了两例 JS 患者中同源隐性功能丧失错义变体,其为轻度 JS。最近,在常染色体显性遗传 COMA 的家族中发现了杂合截断变体,偶尔与轻度 DD 和细微小脑异常相关。
我们重新分析了两个包含 1097 例遗传 JS 基因检测患者的下一代测序(NGS)数据。
在 17 个家系的 22 例患者(1.5%)中检测到杂合截断和剪接位点变体,这些患者具有强烈的男性优势(86%),8 例无症状的父母也携带这种变体。患者表现为 COMA、肌张力减退、共济失调和轻度 DD,仅三分之一表现出不同严重程度的智力障碍。脑 MRI 显示出一致的发现,其特征为蚓部发育不良、上小脑发育不良和上小脑脚细微至轻度异常。在两位接受测试的无症状父母中也观察到了同样的模式。
杂合截断或剪接位点变体引起一种新的神经发育综合征,包括 COMA 和轻度 JS,可能是重叠的实体。变体可以是新生的,也可以从健康的父母那里遗传而来,这代表了具有显性遗传和低外显率的 JS 的首个病因。对这种情况的认识将提高 JS 基因检测的诊断率,并允许对预后、医学监测和复发风险进行适当的咨询。