Translational Medicine and Clinical Pharmacology, Boehringer Ingelheim Pharma GmbH & Co. KG, Birkendorfer Str. 65, 88397, Biberach an der Riss, Baden Württemberg, Germany.
Boehringer Ingelheim Canada Ltd, Burlington, ON, Canada.
Eur J Drug Metab Pharmacokinet. 2022 Jan;47(1):91-103. doi: 10.1007/s13318-021-00723-y. Epub 2021 Oct 29.
Increased glycine availability at the synaptic cleft may enhance N-methyl-D-aspartate receptor signalling and provide a promising therapeutic strategy for cognitive impairment associated with schizophrenia. These studies aimed to assess the pharmacokinetics of BI 425809, a potent glycine-transporter-1 inhibitor, when co-administered with a strong cytochrome P450 3A4 (CYP3A4) inhibitor (itraconazole) and inducer (rifampicin).
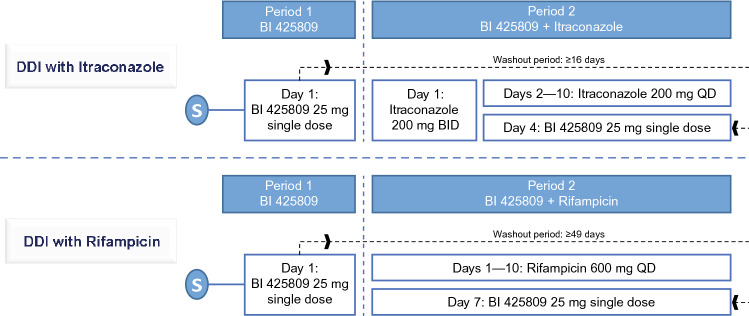
In vitro studies using recombinant CYPs, human liver microsomes, and human hepatocytes were conducted to determine the CYP isoforms responsible for BI 425809 metabolism. In addition, two open-label, fixed-treatment period, phase I studies in healthy male volunteers are described. Period 1: participants received oral BI 425809 25 mg (single dose) on day 1; period 2: participants received multiple doses, across 10 days, of oral itraconazole or rifampicin combined with a single dose of oral BI 425809 25 mg on day 4/7 of the itraconazole/rifampicin treatment, respectively. Pharmacokinetic and safety endpoints were assessed in the absence/presence of itraconazole/rifampicin and included area under the concentration-time curve (AUC) over the time interval 0-167 h (AUC; itraconazole), 0-168 h (AUC; rifampicin), or 0-infinity (AUC; rifampicin and itraconazole), maximum measured concentration (C) of BI 425809, and adverse events.
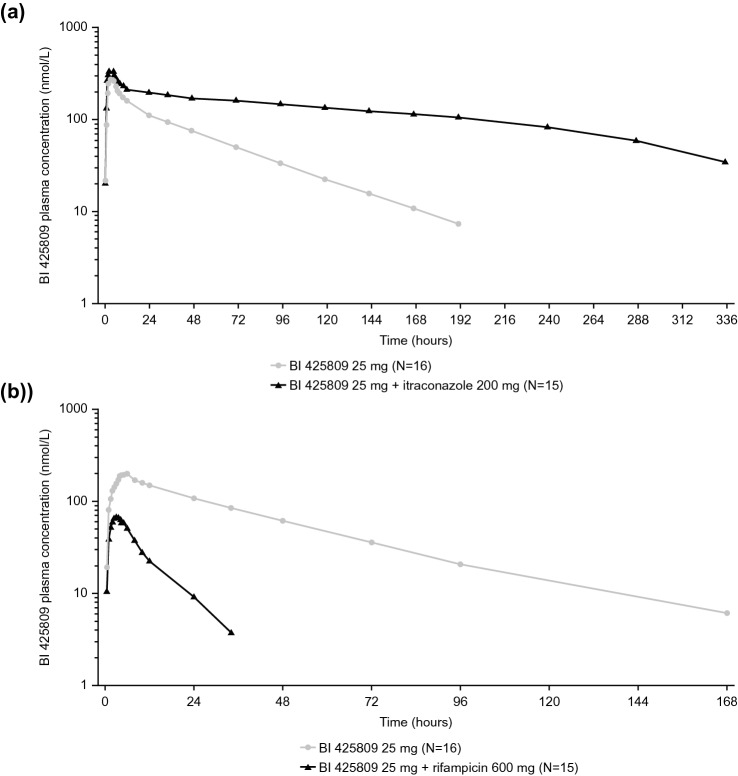
In vitro results suggested that CYP3A4 accounted for ≥ 90% of the metabolism of BI 425809. BI 425809 exposure (adjusted geometric mean ratio [%]) was higher in the presence of itraconazole (AUC: 265.3; AUC: 597.0; C: 116.1) and lower in the presence of rifampicin (AUC: 10.3; AUC: 9.8; C: 37.4) compared with BI 425809 alone. Investigational treatments were well tolerated.
Systemic exposure of BI 425809 was altered in the presence of strong CYP3A4 modulators, corroborating in vitro results that CYP3A4 mediates a major metabolic pathway for BI 425809.
NCT02342717 (registered on 15 January 2015) and NCT03082183 (registered on 10 March 2017).
突触间隙中甘氨酸可用性增加可能会增强 N-甲基-D-天冬氨酸受体信号传递,并为与精神分裂症相关的认知障碍提供有前景的治疗策略。这些研究旨在评估 BI 425809(一种有效的甘氨酸转运体-1 抑制剂)与强细胞色素 P450 3A4(CYP3A4)抑制剂(酮康唑)和诱导剂(利福平)联合给药时的药代动力学。
使用重组 CYP、人肝微粒体和人肝细胞进行体外研究,以确定负责 BI 425809 代谢的 CYP 同工酶。此外,还描述了两项在健康男性志愿者中进行的开放标签、固定治疗期、I 期研究。第 1 期:参与者在第 1 天接受口服 BI 425809 25 mg(单次剂量);第 2 期:参与者分别接受酮康唑或利福平联合口服 BI 425809 25 mg 单次剂量,连续 10 天的多剂量治疗,分别在酮康唑/利福平治疗的第 4/7 天。在没有/存在酮康唑/利福平的情况下评估了药代动力学和安全性终点,并包括 0-167 h 时间间隔内的浓度-时间曲线下面积(AUC;酮康唑)、0-168 h(AUC;利福平)或 0-无穷大(AUC;酮康唑和利福平)、BI 425809 的最大测量浓度(C)和不良事件。
体外结果表明,CYP3A4 占 BI 425809 代谢的≥90%。酮康唑存在时,BI 425809 暴露(调整后的几何均数比 [%])更高(AUC:265.3;AUC:597.0;C:116.1),利福平存在时 BI 425809 暴露更低(AUC:10.3;AUC:9.8;C:37.4)与 BI 425809 单独相比。研究治疗方法耐受性良好。
在强 CYP3A4 调节剂存在的情况下,BI 425809 的全身暴露发生改变,这与 CYP3A4 介导 BI 425809 的主要代谢途径的体外结果一致。
NCT02342717(2015 年 1 月 15 日注册)和 NCT03082183(2017 年 3 月 10 日注册)。