Pigazzini Maria Lucia, Lawrenz Mandy, Margineanu Anca, Kaminski Schierle Gabriele S, Kirstein Janine
Department of Molecular Physiology and Cell Biology, Leibniz Research Institute for Molecular Pharmacology in the Forschungsverbund Berlin e.V. (FMP), Berlin, Germany.
NeuroCure Cluster of Excellence, Charité Universitätsmedizin Berlin, Berlin, Germany.
Front Mol Neurosci. 2021 Oct 15;14:721749. doi: 10.3389/fnmol.2021.721749. eCollection 2021.
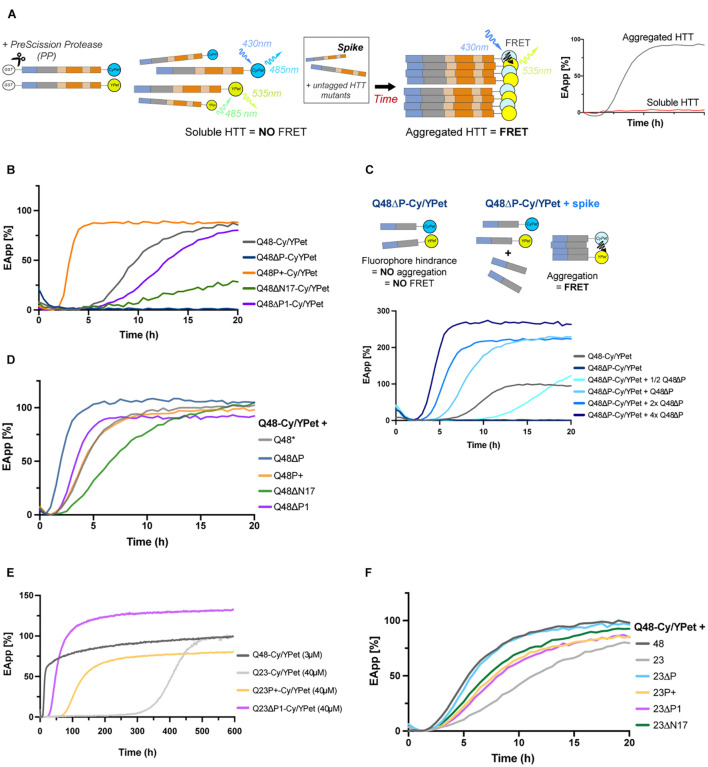
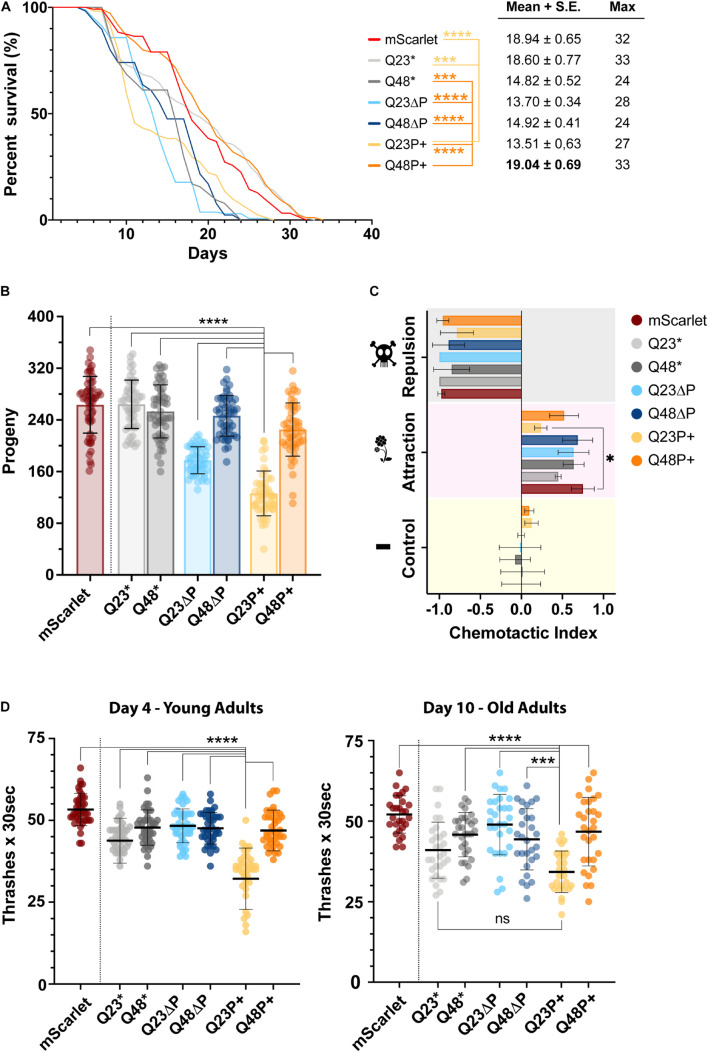
Huntington's disease is a dominantly inherited neurodegenerative disorder caused by the expansion of a CAG repeat, encoding for the amino acid glutamine (Q), present in the first exon of the protein huntingtin. Over the threshold of Q39 HTT exon 1 (HTTEx1) tends to misfold and aggregate into large intracellular structures, but whether these end-stage aggregates or their on-pathway intermediates are responsible for cytotoxicity is still debated. HTTEx1 can be separated into three domains: an N-terminal 17 amino acid region, the polyglutamine (polyQ) expansion and a C-terminal proline rich domain (PRD). Alongside the expanded polyQ, these flanking domains influence the aggregation propensity of HTTEx1: with the N17 initiating and promoting aggregation, and the PRD modulating it. In this study we focus on the first 11 amino acids of the PRD, a stretch of pure prolines, which are an evolutionary recent addition to the expanding polyQ region. We hypothesize that this proline region is expanding alongside the polyQ to counteract its ability to misfold and cause toxicity, and that expanding this proline region would be overall beneficial. We generated HTTEx1 mutants lacking both flanking domains singularly, missing the first 11 prolines of the PRD, or with this stretch of prolines expanded. We then followed their aggregation landscape with a battery of biochemical assays, and in novel models of expressing the HTTEx1 mutants pan-neuronally. Employing fluorescence lifetime imaging we could observe the aggregation propensity of all HTTEx1 mutants during aging and correlate this with toxicity via various phenotypic assays. We found that the presence of an expanded proline stretch is beneficial in maintaining HTTEx1 soluble over time, regardless of polyQ length. However, the expanded prolines were only advantageous in promoting the survival and fitness of an organism carrying a pathogenic stretch of Q48 but were extremely deleterious to the nematode expressing a physiological stretch of Q23. Our results reveal the unique importance of the prolines which have and still are evolving alongside expanding glutamines to promote the function of HTTEx1 and avoid pathology.
亨廷顿舞蹈症是一种常染色体显性遗传的神经退行性疾病,由一种编码谷氨酰胺(Q)的CAG重复序列扩增引起,该重复序列存在于亨廷顿蛋白的第一个外显子中。超过Q39阈值时,亨廷顿蛋白外显子1(HTTEx1)往往会错误折叠并聚集成大的细胞内结构,但这些终末期聚集体或其生成过程中的中间体是否具有细胞毒性仍存在争议。HTTEx1可分为三个结构域:一个N端的17个氨基酸区域、聚谷氨酰胺(polyQ)扩增区域和一个C端富含脯氨酸的结构域(PRD)。除了扩增的polyQ外,这些侧翼结构域也会影响HTTEx1的聚集倾向:N17起始并促进聚集,而PRD则对其进行调节。在本研究中,我们聚焦于PRD的前11个氨基酸,这是一段纯脯氨酸序列,是在不断扩展的polyQ区域中最近进化出来的。我们假设这个脯氨酸区域会随着polyQ一起扩展,以抵消其错误折叠和产生毒性的能力,并且扩展这个脯氨酸区域总体上是有益的。我们构建了单独缺失两个侧翼结构域、缺失PRD的前11个脯氨酸或扩展了这段脯氨酸序列的HTTEx1突变体。然后,我们通过一系列生化分析以及在全神经元表达HTTEx1突变体的新模型中,追踪它们的聚集情况。利用荧光寿命成像技术,我们可以观察到所有HTTEx1突变体在衰老过程中的聚集倾向,并通过各种表型分析将其与毒性相关联。我们发现,无论polyQ长度如何,扩展的脯氨酸序列的存在有利于随着时间的推移保持HTTEx1的可溶性。然而,扩展的脯氨酸仅在促进携带致病性Q48序列的生物体的存活和健康方面具有优势,但对表达生理性Q23序列的线虫极为有害。我们的研究结果揭示了脯氨酸的独特重要性,它们一直在与不断扩展的谷氨酰胺一起进化,以促进HTTEx1的功能并避免病理状态。